Диабетическая невропатия (ДН) является одним из самых распространенных хронических осложнений сахарного диабета (СД) и наиболее часто встречающейся формой невропатии. По данным ряда клинических исследований, распространенность диабетической периферической невропатии (ДПН) составляет 30-50 % среди всех больных СД.
Так, в рамках многоцентрового исследования, проведенного в 118 клиниках Великобритании, были получены данные о распространенности ДН в группе, включившей 6487 больных СД 1 и 2 типов [1]. Среди больных СД 1 типа независимо от продолжительности заболевания распространенность составила в среднем 32 %, среди больных СД 2 типа — 23 %. С увеличением возраста пациентов и временем заболевания СД частота возникновения ДН возрастала. У пациентов со сроком давности СД менее 5 лет распространенность составляла 21 %, а у пациентов со сроком СД более 10 лет — 37 %. В то время как в возрастной группе 20—29 лет ДН диагностировали лишь среди 5 % пациентов, ее распространенность в возрастной группе 70—79 лет составляла уже 44 % [1].Тем не менее в литературе часто присутствуют противоречивые сведения, неоднозначность которых, скорее всего, может быть следствием разноплановой и неадекватной диагностики при выраженном разнообразии клинической симптоматики, отсутствия унифицированных методов выявления периферической невропатии, а также исследований различных контингентов пациентов.
В последнее время мы часто говорим о многофакторном управлении СД 2 типа, подразумевая при этом влияние на различные звенья патогенеза данного заболевания.
Патогенез диабетической невропатии
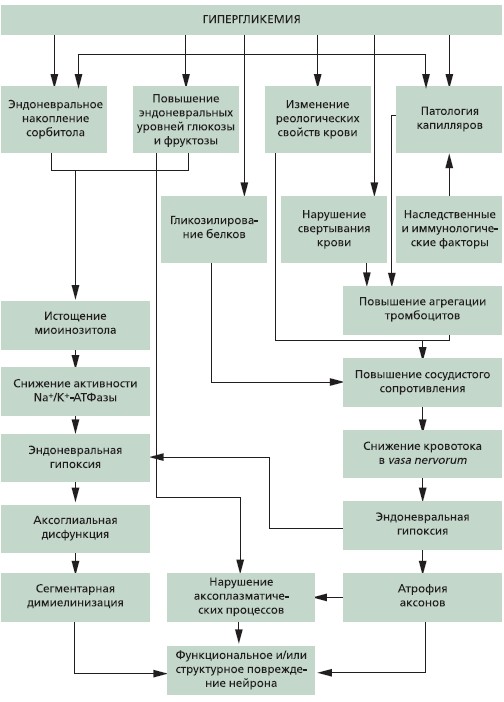
В настоящее время с патогенетической точки зрения ДПН следует рассматривать скорее как комплекс многофакторных событий, в развитии которых центральную роль играет глюкозотоксичность (см. рисунок).
Рисунок.Патогенез диабетической невропатии.
Основные патогенетические механизмы развития ДПН:
- Активизация полиолового пути утилизации глюкозы приводит к накоплению сорбитола и фруктозы, истощению миоинозитола и снижению активности Νа+/К+-АТФазы. Поскольку глюкоза способна проникать в нервную клетку независимо от инсулина, она может неполностью включаться в процесс гликолиза, следствием чего является активация полиолового пути обмена веществ. В результате активизации ключевого фермента альдозредуктазы образуется повышенное количество сорбитола, который диффундирует из нервных клеток или окисляется сорбитолдегидрогеназой с образованием фруктозы.
Именно внутриклеточное накопление сорбитола является причиной различных повреждений нервов. Так, при активации полиолового пути обмена веществ происходит истощение запасов “вторичного мессенджера” миоинозитола.
- Оксидативный стресс — один из ведущих патогенетических механизмов развития ДН, которому в настоящее время уделяется особое внимание. Повышенное образование свободных радикалов, характерное для СД, приводит к нарушению структуры и функций нервных клеток. При оксидативном стрессе в биологической системе увеличивается количество реактивного кислорода или кислородных радикалов. Обычно в организме окислительные и антиоксидантные процессы сбалансированны, поэтому постоянно образующиеся при окислительном стрессе свободные радикалы нейтрализуются. В условиях гипергликемии свободные радикалы образуются в процессе “самоокисления” глюкозы во время образования конечных продуктов ускоренного гликозилирования. Скорость образования свободных радикалов зависит от таковой гликозилирования белков, а следовательно, и от степени гипергликемии. Значение свободных радикалов заключается в их повышенной способности к химическим реакциям. Свободные радикалы вступают в реакции с ненасыщенными соединениями, нарушая структуру ферментных белков и липидов клеточных мембран. На первых стадиях окисления преобладают процессы про- теолиза и активизации перекисного окисления липидов, приводящие к изменению физико-химических свойств биологических мембран. В дальнейшем развивается деструкция липидов мембран, основную роль в которой играют гидролитические ферменты, проникающие из поврежденного аксона. Избыточное образование свободных радикалов с последующим повреждением мембранных структур нейронов и ДНК приводит к нарушению функций нервных клеток. Ткань нерва теряет способность транспортировать инозитол-соединение, необходимое для образования компонентов миелина. Эндоневральная гипоксия, обусловленная патологией микроциркуляции, повышает активность свободных радикалов, которые в свою очередь приводят к тромбогенной трансформации эндотелия сосудистой стенки (посредством активации фактора транскрипции NF-kB) и изменяют структуру стенок капилляров, усиливая эндонев- ральную гипоксию.
Токсичность свободных радикалов ограничивается в присутствии “ловушек радикалов”, поддержание содержания перекисей на низком уровне обеспечивается их участием в метаболических реакциях в качестве интермедиаторов или переводом их в стабильные молекулы. Усиление свободнорадикальных процессов является одним из триггерных механизмов, ведущих к дефициту оксида азота (NO) при СД. Считается, что наиболее ранние нарушения эндотелиальной функции и вазодилатации связаны преимущественно с ослаблением синтеза, высвобождения и/или эффектов эндотелиального NO. Свободные радикалы способны прямо разрушать NO, кроме того, усиление перекисного окисления липидов мембран приводит к повреждению структуры эндотелия и нарушению его NO-продуцирующей способности. Развивается абсолютный или относительный дефицит NO, необходимого для нормальной регуляции сосудистого тонуса. Ослабление NO-зависимых вазодилататорных реакций приводит больных СД к повышению сосудистого тонуса, сужению сосудов с последующим понижением давления кислорода в периферических нервах.
- Нарушения метаболизма n-6 незаменимых жирных кислот и простагландинов, что приводит к изменениям структуры нейрональных мембран, нарушению функции капилляров и реологических свойств крови. Нарушение обмена жирных кислот ведет к нарушениям в циклооксигеназном цикле, снижению продукции вазоактивных субстанций и тем самым к нарушению эндоневраль- ного кровотока.
- Проницаемость барьера кровь/нерв для гликозилированного альбумина, который образуется у пациентов с ДН, может быть повышенной (Poduslo, 1992). Гликозилированные макромолекулы, повышенные концентрации которых отмечаются в эндоневрии, способны посредством прямого токсического эффекта, осмотических изменений или иммунологических механизмов приводить к возникновению невропатии.
- Неферментативное гликозилирование белков основано на способности глюкозы, фруктозы и галактозы вступать в реакции гликозилирования с аминогруппами, входящими в структуры белков, липидов и нуклеиновых кислот. В результате в процессе реакции Майлларда (Maillard) между молекулами сахаров и аминогруппами белков, липидов, ДНК происходит образование обратимых Шиффовых оснований. Из них образуются продукты Амадори (Amadori), создающие конечные продукты гликозилирования (AGE — Advanced glycosylation end products) (Bierhaus, 2004), пройдя через комплексную серию химических превращений. Эти продукты с помощью внутри- и межмолекулярных связей вызывают необратимые изменения структур, в т. ч. белков, и приводят к нарушению их функций. Тут теория гликозилирования белков переплетается со свободнорадикальной теорией: при образовании AGEs в клетке в 50 раз возрастает содержание свободных радикалов.
- Ослабление нейротрофизма, приводящее к снижению экспрессии и истощению нейротрофических факторов, таких как фактор роста нервов, нейротрофин-3 и инсулиноподобный фактор роста, и нарушению аксонального транспорта.
- Иммунные реакции с образованием аутоантител к блуждающему нерву, узлам симпатического ствола и мозговому веществу надпочечника (Rabinowe, 1990), а также воспалительные процессы.
C-пептид. Снижение соотношения инсулин/С-пептид рассматривается в качестве возможного патогенетического пути развития ДПН. Доклинические исследования показали широту физиологического действия С-пептида, а именно: влияние на активность Νа+/К+-АТФазы, эндотелиальной NO-синтетазы, экспрессию нейротрофных факторов, регулирование молекулярных механизмов, лежащих в основе дегенерации нервов у пациентов с СД 1 типа, влияние на взаимодействие факторов транскрипции с ДНК и влияние на явления апоптоза. В исследовании пациентов с СД 1 типа, на протяжении 12 недель получавших подкожные инъекции С-пептида, было выявлено значимое улучшение скорости проведения импульса по чувствительным волокнам икроножного нерва, улучшение вибрационной чувствительности без изменений температурной чувствительности. Эффективность С-пептида в лечении ДПН при СД 2 типа сомнительна и не доказана, что требует дополнительных исследований в этой области [2—6].
ДПН является прогрессирующим заболеванием, которое в отсутствие адекватной терапии приводит к развитию “синдрома диабетической стопы” (ДС). Таким образом, можно сказать, что ДН — сопутствующее СД патологическое состояние, вызывающее повышенную заболеваемость и смертность пациентов. В системе здравоохранения расходы на лечение пациентов, страдающих ДН, исчисляются 1 млрд евро. Высокая стоимость лечения ДН обусловлена прежде всего несвоевременной диагностикой, т. к., как правило, выявляют ДН уже на стадии необратимых изменений и клинически выраженной симптоматики. Старт терапии ДН, возможно, необходимо начинать задолго до появления ее первых симптомов.
Существует традиционная точка зрения, будто СД приводит к развитию ДН после многих лет устойчивой гипергликемии. Однако, по некоторым литературным данным, у 20 % пациентов и более с впервые выявленным СД по результатам электрофизиологического исследования диагностирована ДН, в то время как диабетические ретинопатия и нефропатия практически отсутствовали. Идиопатическая невропатия, по мнению ряда авторов, является маркером предиабета [6]. Эксперименты на лабораторных животных и человеке показали, что транзиторная гипергликемия увеличивает спонтанный разряд от малого диаметра ноцицептивных афферентных С-волокон и клинически связана с повышением невропатической боли [7, 8].
Интересные данные получены в исследовании Dan Ziegler и соавт., проведенном в 2009 г. в Институте клинической диабетологии (Лейбниц). Целью исследования стало определение распространенности и факторов риска развития невропатической боли у пациентов с СД, нарушением гликемии натощак (НГН), нарушением толерантности к глюкозе (НТГ) или нормальной толерантностью к глюкозе. В исследовании приняли участие 393 добровольца с наличием симптомов нейропатии. Исследуемая популяция была поделена на 2 группы: пациенты с наличием СД (n = 195) и группа контроля (n = 198). Обе группы были сопоставимы по возрасту и полу. В контрольной группе 44 (23,9 %) больных имели сочетание НТГ и НГН; из них у 71 (35,9 %) пациента выявлено НГН, у 81 — НТГ. Распространенность (95 % доверительный интервал) невропатической боли составила 13,4 % (8,9—18,3) для больных СД, 8,7 % (2,4— 20) — для лиц с НТГ и 1,2 % (0,03—6,7) — для пациентов с НГН (p = 0,0003). Во всей популяции (n = 393) возраст, масса тела, заболевания периферических артерий, СД считались факторами риска, в значительной степени ассоциированными с невропатической болью, в отличие от группы с СД, где таковыми были только возраст, масса тела и заболевания периферических артерий. Таким образом, распространенность невропатической боли увеличилась в 2—3 раза у пациентов с наличием НТГ и СД по сравнению с группой НГН. За исключением диабета, преобладающими факторами риска можно назвать возраст, ожирение и заболевания периферических артерий.
Кроме того, в исследовании впервые продемонстрировано, что ДН является независимым фактором, ассоциированным с окружностью талии и заболеваниями периферических артерий. Так, было показано, что увеличение окружности талии на 1 см связано с увеличением вероятности развития невропатии на 4 % [9]. Однако полученные результаты не подтверждают данных других исследований [10].
Согласно ряду авторов, 25—62 % больных идиопатической невропатией страдают предиабетом; среди них периферическая невропатия развивается у 11—25 %, а у 13—21 % — невропатические боли. Наиболее чувствительный тест для оценки состояния метаболизма глюкозы — это оральный глюкозотолерантный тест (ОГТТ). Лица с предиабетом имеют менее серьезные осложнения, чем пациенты с уже манифестировавшим СД. Основными патогенетическими механизмами развития невропатии являются гипергликемия, микрососудистые осложнения, дислипидемия и метаболический синдром. Сенсорные нарушения при ДН выявляются раньше моторных нарушений, и их достаточно легко обнаружить. Диагноз ДН должен опираться на тщательное клиническое и электрофизиологическое обследования. ОГТТ следует проводить всем пациентам с наличием идиопатической невропатии [11].
Интерес к нейрометаболической терапии неврологических осложнений СД вызывает большой интерес как у клиницистов, так и у физиологов. В последнее время появились новые клинические и экспериментальные данные, обосновывающие применение этой группы препаратов у пациентов с диабетической полиневропатией.
Известные механизмы действия Актовегина вызвали интерес к его использованию у больных СД, т. к. при этом заболевании клеточная патология связана и с метаболическими нарушениями, и с изменением кровотока в системе микроциркуляции [12, 13]. Основные исследования по оценке эффективности Актовегина проведены при диабетическом поражении периферических нервов — диабетической дистальной симметричной сенсорномоторной полиневропатии (ДПН) [14—16].
W.Jansen и E.Beck изучили действие перорального приема Актовегина у больных СД с ДПН в рамках контролируемого исследования: одна группа из 35 больных получала плацебо, другая группа из 35 больных — таблетки Актовегина (по 600 мг 3 раза в сутки) в течении 24 недель [16]. Критериями оценки эффективности препарата служили клинические характеристики полиневропатии (сухожильные рефлексы, поверхностная и глубокая чувствительность, интенсивность болевого синдрома) и электромиографические показатели функции периферических нервов (скорость распространения возбуждения, а также расстояние, которое больные могли пройти без боли). Улучшение состояния больных в группе лечения Актовегином отмечено у большинства пациентов через 8 недель после начала лечения, а оптимальный эффект достигался через 16 недель лечения. Показано достоверное улучшение на фоне лечения Актовегином по сравнению с группой плацебо практически всех клинических показателей: расстояния ходьбы без боли, сухожильных рефлексов, поверхностной и глубокой чувствительности (р < 0,01). Скорость распространения возбуждения достоверно (р < 0,001) увеличивалась в группе лечения по сравнению с группой плацебо. Пациенты группы лечения чувствовали себя лучше и предъявляли меньше жалоб на нарушение психоэмоционального состояния, что коррелировало с улучшением их физического состояния.
В 2009 г. опубликованы результаты рандомизированного двойного слепого плацебо-контролируемого исследования лечения больных СД 2 типа с ДПН препаратом Актовегин, которое проводилось в 26 клинических центрах России, Украины, Казахстана. Оценивали клиническую эффективность и безопасность Актовегина у пациентов с СД 2 типа и клиническими проявлениями ДПН. Всего в исследование были включены 567 больных: 281 больному проведено сначала 20 внутривенных инфузий Актовегина (250 мл 20 % раствора), а затем в течение 140 дней пациенты получали драже Актовегина по 600 мг 3 раза в сутки. Внутривенную плацебо-терапию, а затем терапию драже с плацебо получали 286 больных СД с ДПН. Основными критериями эффективности препарата в этом исследовании были жалобы больных на онемение, боль, жжение, покалывание иголками, которые оценивали по шкале невропатических нарушений, и порог вибрационной чувствительности, который определялся в нескольких точках на ногах с помощью биотензиометра. Вторичными критериями эффективности служили шкала неврологических симптомов и показатели качества жизни. Наилучшие результаты были отмечены в отношении неприятных ощущений больных в ногах, причем улучшение отмечалось как по суммарной оценке всех симптомов, так и в отношении каждого конкретного симптома. Выявлено достоверное уменьшение сенсорного неврологического дефицита. Уменьшение порога вибрационной чувствительности было высоко достоверным при использовании Актовегина по сравнению с плацебо. В течение всего исследования проводилось определение уровня глюкозы натощак и гликированного гемоглобина. Полученные результаты свидетельствуют о том, что эффективность Актовегина связана с действием препарата, а не с изменением контроля СД. В исследовании не отмечено значимых побочных явлений. Показано значимое улучшение качества жизни (по шкале психического здоровья) в группе Актовегина по сравнению с плацебо. Отмечено, что группы пациентов, получавших Актовегин и плацебо, имели сравнимый профиль безопасности. Американский фармкомитет (FDA — Food and Drug Administration) разработал определенные критерии для лекарственных средств, которые могут быть зарегистрированы как препараты для лечения ДПН:
- действие на патогенетические механизмы,
- уменьшение симптомов невропатии,
- улучшение функции нерва,
- отсутствие значительных побочных эффектов,
- уменьшение риска гибели нервных волокон.
Проведенные исследования позволяют считать, что Актовегин соответствует большинству из этих критериев и может быть использован для лечения ДПН [17, 18].
Резюмируя все вышеизложенное, можно сказать, что только при своевременной диагностике и ранней инициации терапии возможен успех в достижении цели лечения ДН. Кроме того, существует необходимость в дальнейших исследованиях для более точного определения эпидемиологической связи между НТГ и идиопатической невропатией с целью разработки определенных мер, направленных на предотвращение развития более серьезных осложнений СД — ампутаций нижних конечностей.

{kind=link}