
Артериальная гипертензия (АГ) остается одним из главных факторов риска ремоделирования сердца (гипертрофия миокарда) и сосудов (увеличение толщины внутреннего слоя с сужением их просвета, эндотелиальная дисфункция), развития сердечно-сосудистых заболеваний и осложнений, таких как атеросклероз, ишемическая болезнь сердца, инфаркт миокарда, сердечная недостаточность (СН) – часто с сохраненной фракцией выброса.
Полагают, что при подобных изменениях в сердечно-сосудистой системе задействованы многие механизмы, в частности активация ренин-ангиотензин-альдостероновой системы (РААС), активация симпато-адреналовой системы, увеличение объема циркулирующей жидкости и нарушения электролитного баланса. Следует сказать, что эти системы тесно взаимосвязаны и активация одного звена приводит к изменению функционирования других звеньев, участвующих в повышении артериального давления (АД) и структурно-функциональном ремоделировании органов-мишеней.
Для лечения повышенного АД используют восемь классов лекарственных препаратов, воздействующих на разные патогенетические звенья АГ, причем более трех четвертей пациентов с АГ требуют комбинированной терапии [1].
В соответствии с современными позициями по лечению пациентов с АГ в большинстве случаев рекомендуют использовать комбинации ингибиторов РААС с антагонистами кальция или с диуретиками. Препаратам, ингибирующим активность РААС, – ингибиторам ангиотензинпревращающего фермента (иАПФ) и сартанам, придают особое значение в связи с тем, что за последние годы наши представления о системах, регулирующих структурно-функциональное состояние сердца и сосудов, претерпели существенные изменения, были выявлены новые механизмы функционирования РААС и пути, по которым эти препараты оказывают влияние на РААС [2, 3].
В настоящее время описано три разновидности ренин-ангиотензиновой системы (РАС): это классическая гормональная РААС; тканевые РАС, расположенные на поверхности клеток, и внутриклеточные РАС.
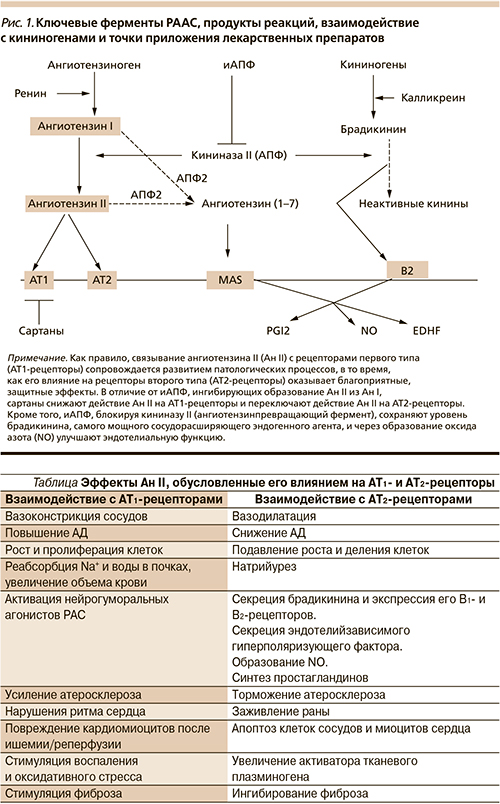
Классическая РААС – одна из главных регуляторных систем организма, участвует в поддержании различных жизненно важных показателей в реакциях немедленного типа: контролирует АД, ионный гомеостаз, уровень ионов Na+, водный баланс, объем крови, а также функциональную активность почек. Ключевые ферменты РААС, продукты реакций, взаимодействие с калликреиновой системой и точки приложения лекарственных препаратов показаны на рис. 1.
Система тканевых, или локальных, РАС участвует в хроническом действии Ан II на клетки, которое не всегда связано с регуляцией АД, а вызывает разнообразные клеточные ответы, часто заканчивающиеся цитотоксическим эффектом.
Для этой системы характерны две особенности:
- Благодаря фиксации ренина и АПФ на поверхности клетки локально образуются высокие концентрации Ан II, изменяющие активность клеток-мишеней, в которых экспрессированы рецепторы для Ан II.
- Образовавшийся Ан II действует аутокринно, т.е. на клетку-продуцент, или паракринно – на окружающие клетки.
Система внутриклеточных РАС находится в цитоплазме некоторых видов клеток (кардиомиоцитах, адипоцитах, клетках почек), содержит все или, во всяком случае, ключевые компоненты РАС: ферменты, субстраты и продукты реакций, а также АТ1- и АТ2-подобные рецепторы, экспрессированные на лизосомах [4], митохондриях (МХ) [5], ядрах клеток [6, 7], и участвуют в фагоцитозе, энергетическом обмене и активации генов, продукты которых контролируют гипертрофию клеток, сократительную активность сердца и другие процессы (см. таблицу).
Интракринные эффекты внутриклеточного Ан II тесно связаны с изменением уровня одного из главных вторичных мессенджеров – ионов кальция (Са2+) в цитоплазме, который увеличивается в результате выхода Са2+ из внутриклеточных Са2+-депо (SR-депо) в цитоплазму (Е.И. Асташкин, Глезер, 2012).
Установлено, что выход Са2+ из внутриклеточных депо происходит по Са2+-каналам, формируемым рецепторами для инозитол трисфосфата (IP3). Об этом свидетельствует следующий факт: блокада IP3-рецепторов Са2+-депо подавляет действие интракринного Ан II: блокирует выход Са2+ из депо и тормозит Са2+-зависимую активацию генов, связанных с Са2+-зависимым фактором транскрипции генов NF-kB [6].
Активация АТ1- и АТ2-подобных рецепторов, локализующихся на внутренней мембране МХ [5], изменяет потребление МХ кислорода и тем самым влияет на синтез АТФ. Влияние Ан II на АТ1-рецепторы в МХ может прямо (в результате воздействия на ферментативные комплексы дыхательной цепи) или опосредованно (например, через поступление ионов Са2+ в матрикс МХ) изменять дыхание этих органелл. Жирорастворимые сартаны подавляют эффекты Ан II, связанные с активацией АТ1-рецепторов МХ [5], и восстанавливают картину экспрессии АТ1- и АТ2-рецепторов, наблюдаемую на молодых животных [4, 5, 7].
Показано, что в изолированных кардиомиоцитах экзогенный Ан II, введенный в клетку, связывался с ядром и активировал фактор транскрипции генов NF-kB. Этот процесс блокировался внутриклеточным введением валсартана, действующим на АТ1-подобные рецепторы ядра, но не на АТ2-рецепторы [6].
Следует отметить, что внутри группы как ингибиторы АПФ, так и сартаны существенным образом отличаются друг от друга. Они имеют разную химическую структуру, обладают различными физико-химическими свойствами, различаются по растворимости в воде и жирах. Это влияет на способность препаратов проникать в клетки и клеточные органеллы через липидный бислой мембран. Благодаря этому жирорастворимые иАПФ и сартаны могут не только блокировать активность АПФ и АТ1-рецепторов классической эндокринной РААС и тканевой поверхностной РАС, но и влиять на внутриклеточную РАС, изменяя интракринный характер действия Ан II [8].
Самое большое число исследований в этой области проведено с жирорастворимым препаратом валсартан.
У валсартана (Диован) из всех препаратов этой группы самый широкий спектр зарегистрированных показаний к применению: АГ, хроническая СН (II–IV функциональные классы по классификации NYHA – New York Heart Association) в составе комплексной терапии, повышение выживаемости пациентов с острым инфарктом миокарда, осложненным левожелудочковой недостаточностью и/или систолической дисфункцией левого желудочка, при наличии стабильных показателей гемодинамики.
Проведена масса исследований эффективности валсартана в отношении разных категорий пациентов с АГ. Его антигипертензивный эффект связан, с одной стороны, с блокадой АТ1-рецепторов, с другой – со стимуляцией АТ2-рецепторов под влиянием Ан II, а также опосредуется через образование Ан 1-7 и АПФ2, что сопровождается увеличением содержания брадикинина, который, как уже указывалось, стимулирует продукцию NO и тем самым увеличивает содержание циклического гуанозинмонофосфата (сGMP). Это приводит к улучшению эндотелиальных свойств сосудов, их расширению, снижению периферического сопротивления сосудов, снижению АД.
Валсартан входит в состав фиксированных комбинированных готовых лекарственных форм, содержащих амлодипин (препарат Эксфорж – амлодипин + валсартан) или амлодипин и гидрохлоротиазид (КО Эксфорж). Эти препараты широко и успешно используются в клинической практике [9, 10].
Помимо антигипертензивного эффекта валсартан обладает выраженными органопротективными свойствами: уменьшает выраженность гипертрофии миокарда, оказывает положительные эффекты, предупреждая постинфарктное ремоделирование сердца (исследование VALIANT) [11] и улучшая течение СН (исследование Val-He-FT) [12].
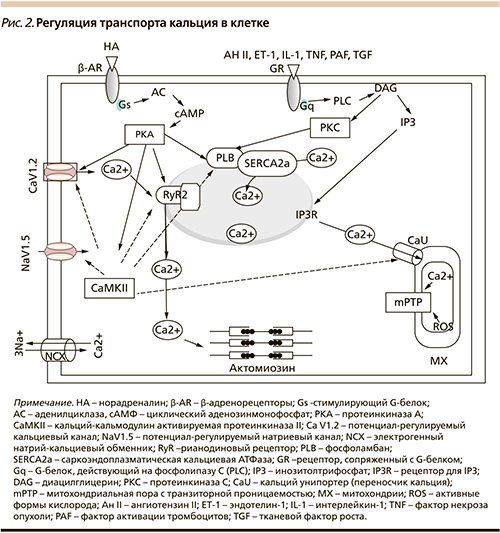
За последнее время получены важные данные о новых механизмах, по которым валсартан оказывает эти положительные эффекты. Многие из этих эффектов связаны с влиянием валсартана на интракринную РАС и обусловлены изменениями обмена в кардиомиоцитах или гладкомышечных клетках Ca2+. В связи с этим рассмотрим основные процессы обмена Cа2+ в кардиомиоцитах, схематично представленные на рис. 2.
Регуляция сократительной активности кардиомиоцитов в физиологических условиях
Как известно, сокращение миоцитов сердца в фазу систолы запускается транспортом небольших количеств ионов кальция (Са2+) через потенциал-регулируемые медленные Са2+-каналы сарколеммы (L-VOC- каналы) в цитоплазму при деполяризации мембраны [13]. Этот триггерный Са2+ служит сигналом для выхода больших количеств ионов Са2+ из внутриклеточного Са2+-депо саркоплазматического ретикулума (SR-депо) по кальциевым каналам, формируемым рианодиновыми рецепторами (RyR). Именно этот Са2+ используется актомиозиновым комплексом при мышечном сокращении.
Различные внеклеточные стимулы, например стимуляция β-адренорецепторов кардиомиоцитов норадреналином [14], увеличивают активность L-VOC-каналов вследствие стимуляции аденилатциклазной системы. Увеличение уровня циклического аденозинмонофосфата (сАМР) активируется сАМР-зависимой протеинкиназой А (PKA). Этот фермент фосфорилирует различные мишени, включая L-VOC-каналы, RyR Са2+-депо, ферменты, контролирующие Са2+-цикл и мышечное сокращение [15].
В фазу диастолы ионы Са2+ удаляются из цитоплазмы через плазматическую мембрану с помощью Са2+-АТФазы и Na+/Са2+-обменника, а также в результате активного транспорта ионов Са2+ в SR-депо, осуществляемого Са2+-АТФазой депо (Са2+-АТФаза сарко-эндоплазматического ретикулума – SERCA2а), что необходимо для последующего сокращения.
Активность SERCA2a подавляется фосфоламбаном (PLB). При фосфорилировании PLB активность SERCA2a возрастает. Уменьшение фосфорилирования PLB может быть обусловлено снижением активности и экспрессии PKA и стимуляцией фосфатазы РР1-α, которые фосфорилируют и дефосфорилируют PLB соответственно [16, 17].
Следует отметить, что в качестве резервного депо для ионов Са2+ выступают МХ, перегрузка которых ионами Са2+ приводит к гибели кардиомиоцитов.
Снижение выведения ионов Са2+ из цитоплазмы в фазу диастолы вызывает постепенное накопление ионов Са2+ в клетке, активирует процессы, лежащие в основе их гипертрофии и ремоделирования сердца, затрудняет расслабление кардиомиоцитов, что ведет к развитию диастолической СН.
Ионы Са2+ в клетке влияют на активность Са2+-связывающих белков, участвующих в регуляции разнообразных ферментативных каскадов. Регуляторные биохимические эффекты Са2+ в клетках реализуются в результате его связывания с кальмодулином (Ca2+/CaM) и воздействием этого комплекса на кальций-кальмодулинзависимую протеинкиназу (СаМРКII), которая фосфорилирует различные белки. Удаление фосфатных групп (дефосфорилирование) происходит под влиянием другого Са2+-зависимого фермента – кальцинеурина, который является серин-треониновой фосфатазой, активируется Ca2+/CaM и воздействует на ядерный фактор транскрипции генов – NF-kB. Другим важным Са2+-регулируемым белком является кальпаин – член большого семейства цитозольных Са2+-активируемых цистеиновых протеаз. При активации ионами Са2+ эта протеаза разрушает клеточные мембраны и разнообразные внутриклеточные белки и изменяет их активность. Таким образом, действие кальпаина необратимо связано с процессами дегенерации и апоптоза клеток.
Нарушение поступления и выведения Са2+ в цитоплазме (Са2+-цикл) может происходить на любом его этапе – при дефекте L-VOC-каналов, изменении структуры RyR, торможении активности Са2+-АТФаз и Nа+/Са2+-обменника, дисфункции кальмодулина, СаМРКII, кальцинеурина.
Патофизиологические условия, способствующие развитию гипертрофии миокарда и СН
Длительная активация эндокринной и тканевых РАС, сопровождающаяся пролонгированным образованием больших количеств Ан II, и стимуляция симпатической нервной системы с повышенным выделением из нервных окончаний норадреналина активируют внутриклеточные сигнальные процессы и гены, продукты которых контролируют рост клеток. Конечным итогом этих процессов служит развитие гипертрофии миокарда.
При гипертрофии миокарда меняются не только структура, размеры и форма камер сердца, но и состояние ферментов (метаболическое ремоделирование), контролирующих в т.ч. энергетический обмен, активный транспорт ионов в кардиомиоцитах, их электрические характеристики и процессы мышечного сокращения [13].
Установлено, что между нарушениями в кардиомиоцитах активности различных типов АТФаз, ответственных за потоки ионов через мембраны за счет энергии гидролиза АТФ (активный транспорт ионов), изменением ионного гомеостаза и сократительной активностью кардиомиоцитов существует тесная взаимосвязь [18].
Снижение активности Na+/K+-АТФазы, Са2+-АТФазы плазматической мембраны и SR-депо (SERCA2a), повышение активности Na+/Са2+-обменника наружной мембраны при гипертрофии миокарда тормозят активный транспорт ионов Na+, K+, Са2+, снижают ионные градиенты через наружную и внутриклеточные мембраны, прежде всего саркоплазматического ретикулума (SR-депо ионов Са2+). Это сопровождается увеличением уровней Na+ и Ca2+ в цитоплазме, а также приводит к поступлению ионов Са2+ в матрикс МХ [13, 19]. Нарушения ионного гомеостаза нарушают электромеханическое сопряжение в миоцитах сердца и вызывают формирование диастолической, а затем и систолической недостаточности [20–22].
Нарушению транспорта ионов способствует также снижение активности PKA, экспрессии образования фосфорилированного PLB (p-PLB) при возрастании активности протеин фосфатазы1-α (РР1-α).
Активация РАС и развитие гипертрофии сердца сопровождаются увеличением экспрессии Са2+ регулирующих белков – кальпаина I, экспрессии и активности кальцинеурина, а также двух изоформ СаМРКII – СаМРКII-δA и СаМРК II-δВ, при снижении уровней мРНК и белка изоформы СаМРК II-δС [16, 17, 23].
Действие валсартана на патологические изменения при гипертрофии и СН
Снижение выраженности гипертрофии под влиянием валсартана может быть обусловлено прямым блокирующим эффектом действия Ан II на АТ1-рецептор и уменьшением выраженности эффектов норадреналина [24].
Действие сартанов на АТ1-рецепторы приводит к коррекции активности кальциевого цикла как на уровне Са2+-каналов наружной мембраны клетки, так и в SR-депо, что сопровождается улучшением сократимости кардиомиоцитов. Блокада взаимодействия Ан II с АТ1-рецепторами подавляет снижение активности SERCA2 при СН и восстанавливает регуляцию внутриклеточного уровня ионов Са2+ в SR-депо.
Получены также данные, согласно которым валсартан может оказывать благоприятное действие на структурно-функциональное состояние сердца при СН, вызванное перегрузкой объемом и давлением. Валсартан в этих условиях ингибировал сигнальные пути, связанные с кальпаином I, кальцинеурином, экспрессией и активностью двух изоформ СаМРКII-δ: СаМРКII-δA и СаМРКII-δВ, уменьшал выраженность гипертрофии миокарда [26]. Сходные данные были получены в другом исследовании, где валсартан улучшал функциональную активность сердца у кроликов с СН в результате снижения экспрессии белка СаMPKII и активности этого фермента [27].
Валсартан повышает активность PKA и снижает активность протеинфосфатазы 1-α (фосфатазы РР1-α) (18), что сопровождается увеличением уровня фосфорилированного фосфоламбана ser16-р-PLB, а это увеличивает активность SERCA2a и транспорт Са2+ в SR-депо. При этом валсартан предупреждает развитие ремоделирования левого желудочка и улучшает систолическую дисфункцию, вызываемую длительной перегрузкой объемом и давлением.
При гипертрофии, индуцированной норадреналином, валсартан повышает активность исходно сниженных АТФазы миозина, Na+/K+-АТФазы и Са2+-АТФаз плазматической мембраны и SR-депо, что восстанавливает градиенты ионов Na+, K+ и Са2+, благоприятно влияет на электрическую активность сердца и усиливает сократительную активность миокарда.
При экспериментальной СН, вызванной перегрузкой объемом и увеличенной частотой сердечных сокращений, валсартан увеличивал сниженную активность SERCA2 [18] и, соответственно, активный транспорт Са2+ в SR-депо, снижал высокий уровень Са2+ в цитоплазме во время диастолы, который при СН был обусловлен неконтролируемым выходом Са2+ из SR-депо, восстанавливал стехиометрию и состав компонентов макромолекулярного комплекса RyR2 [16]. Все эти процессы улучшали Са2+-цикл и функциональную активность кардиомиоцитов.
Одно из объяснений такого защитного действия валсартана связано с его влиянием на АТ1-рецепторы в пресинаптической мембране симпатических нейронов, окончания которых иннервируют миокард, в результате чего через пресинаптическую мембрану симпатических нервов меньше секретируется норадреналина и возрастает его обратный захват в нейроны, что увеличивает внутриклеточный пул норадреналина. Это снижает поступление норадреналина к кардиомиоцитам, уменьшается величина адренергического стимула, влияющего на β-адренорецепторы кардиомиоцитов и, соответственно, активация β-адренорецепторов. Это тормозит аденилатциклазу, снижает синтез сАМР и уменьшает активность сАМР-зависимой PKA, фосфорилирующей RyR. Падение при действии валсартана фосфорилирования димерного комплекса RyR2 усиливает связывание ряда белков, включая белок FK BP16.6 с RyR2, что способствует стабилизации закрытого состояния Са2+-каналов этого рецептора и уменьшает выход Са2+ из SR-депо в цитоплазму во время диастолы. Это улучшает расслабление кардиомиоцитов, сопряжение между электрической активностью и сократимостью кардиомиоцитов.
Таким образом, валсартан в какой-то мере имитирует действие блокаторов β-адренорецепторов. Только в случае валсартана его защитный эффект обусловлен снижением уровня биогенных аминов, что ослабляет их действие на β-адренорецепторы, в то время как сами блокаторы β-адренорецепторов препятствуют взаимодействию биогенных аминов с рецепторами. Конечный эффект – образование сАМР: снижается при действии как валсартана, так и β-адреноблокаторов, что уменьшает гиперфосфорилирование RyR2 и восстанавливает их нормальную активность.
Совокупность этих результатов позволяет сделать следующее заключение: валсартан эффективно подавляет экспрессию генов, продукты которых играют центральную роль во внутриклеточной Са2+-регуляции разнообразных клеточных процессов, контролирующих не только гипертрофию сердца, но и его ремоделирование, электромеханическое сопряжение и мышечное сокращение.
Важным аспектом действия валсартана служит влияние на фиброз и апоптоз кардиомиоцитов. Длительное действие Ан II и норадреналина стимулирует также деление фибробластов – основных источников коллагена, отложение которого в соединительной ткани лежит в основе развития фиброза сердца и развития нарушения диастолического расслабления. Кроме того, происходит активация генетически запрограммированной клеточной гибели – апоптоза, и суммарное число кардиомиоцитов снижается, что нарушает сократительную функцию миокарда. Фиброз и апоптоз являются теми процессами, которые переводят компенсированную гипертрофию в некомпенсированную, т.е. ее патологическую форму.
Валсартан снижает содержание коллагена в миокарде и уменьшает фиброз сердца. Это очень важный вид активности валсартана, поскольку фиброз сердца не только связан с переходом от компенсированной гипертрофии к СН, но и вызывает аритмии, случаи внезапной сердечной смерти и различные серьезные осложнения [24, 25]. Интерстициальная ткань в сердце отвечает за функциональную интеграцию кардиомиоцитов и структурную целостность сердца как органа, что способствует скоординированному ответу клеток и контролирует силу суммарного мышечного сокращения. Способность валсартана нейтрализовывать систолическую и диастолическую дисфункции левого желудочка, а также снижать АД, обусловлена в т.ч. уменьшением содержания коллагена во внеклеточной ткани сердца.
Валсартан снижает развитие апоптоза в гипертрофированном сердце, о чем свидетельствует восстановление индекса мышечной массы сердца под влиянием валсартана.
Влияние валсартана на нарушения активности МХ
Глубокие изменения энергетического обмена в кардиомиоцитах являются одной из главных причин изменения функциональной активности миокарда при различных сердечно-сосудистых заболеваниях [28]. Основным источником синтеза АТФ в сердце остаются МХ, активность которых зависит от ряда факторов и процессов. Неблагоприятное влияние на клетки оказывает воздействие различных внеклеточных факторов, включая некоторые лекарственные препараты, которые при длительном применении подавляют активность МХ и приводят к развитию кардиомиопатий.
Описаны защитные эффекты валсартана при действии цитотоксических лекарственных препаратов, подавляющих активность МХ, в частности широко используемого для противоопухолевой терапии цитостатика доксирубицина [29, 30], который в определенные фазы клеточного цикла прямо взаимодействует, подавляет активность и синтез ДНК.
Одной из главных причин кардиомиопатии, вызываемой доксорубицином, является поражение МХ, в которых увеличивается образование радикалов кислорода; накапливаются ионы Са2+ в просвете МХ; развивается энергетический дисбаланс, связанный с торможением синтеза АТФ и возрастанием скорости его потребления. В экспериментальных моделях кардиомиопатии, вызванной доксирубицином, валсартан снижал набухание МХ и образование свободных радикалов кислорода, а также увеличивал мембранный потенциал на внутренней мембране МХ и синтез АТФ. В результате частичного восстановления структуры и функциональной активности сердца валсартан оказывал защитное действие при кардиомиопатии, индуцированной доксорубицином. Эти результаты показывают, что МХ выступают как одна из главных мишеней действия не только доксорубицина, но и валсартана [29].
Принимая во внимание, что на внутренней мембране МХ экспрессируются АТ1- и АТ2-подобные рецепторы [5], можно предположить, что защитные эффекты валсартана в той или иной степени связаны с его влиянием на МХ, в т.ч. при острой ишемии. При острой ишемии валсартан восстанавливает функциональную активность МХ в кардиомиоцитах в результате сохранения структуры этих органелл, увеличивает мембранный потенциал на внутренней мембране МХ, усиливает потребление кислорода и фосфорилирование АДФ, что приводит к увеличению синтеза АТФ и подавляет механизмы, вызывающие набухание и разрушение МХ, а затем и кардиомиоцитов [31].
Влияние валсартана на инсулиновую резистентность адипоцитов. Роль тканевых макрофагов
Известно, что ингибиторы АПФ и особенно сартаны снижают риск развития сахарного диабета у пациентов с АГ [32, 33]. Снижению инсулинорезистентности при действии этих препаратов придают большое значение в восстановлении нарушенной эндотелиальной функции при АГ, ожирении, сахарном диабете. Влияние на инсулинорезистентность пытались объяснить действием некоторых сартанов на систему РРАR. В настоящее время установлено, что валсартан, не взаимодействующий с РРАR, может уменьшать инсулинорезистентность жировых клеток. Было установлено, что валсартан увеличивал в присутствии макрофагов чувствительность адипоцитов к инсулину [34]. Подобный инсулин-сенсибилизирующий эффект валсартана воспроизводился не только на лабораторных моделях, но и в клинических условиях.
Валсартан вызывал полное восстановление процесса фосфорилирования субстрата-1 инсулинового рецептора и протеинкиназы В (Akt протеинкиназы) и чувствительности адипоцитов к действию инсулина. Более того, валсартан выраженным образом подавлял эндотоксин-индуцированную продукцию цитокинов воспаления, включая интерлейкин-1β (ИЛ-1β), ИЛ-6 и фактор некроза опухоли α, а также активацию ядерного фактора NF-kB и фосфорилирования терминальной киназы c-Jun. Защитный эффект валсартана также наблюдался в макрофагах с дефектом АТ1-рецепторов в наружной мембране и блокадой активности фактора транскрипции генов PPARγ. Эти результаты свидетельствуют о том, что валсартан подавляет воспалительный ответ макрофагов в обход АТ1-рецепторов плазматических мембран и активации внутриклеточного PPARγ. Очевидно, что мишень такого защитного действия валсартана находится внутри клеток [34].
Таким образом, функциональная активность РАС осуществляется на разных уровнях организации в рамках эндокринной РААС; тканевых РАС, экспрессированных на поверхности клеток-мишеней и тканевых внутриклеточных РАС. Валсартан не только корректирует эффекты, связанные с АТ1-рецепторами, экспрессированными на плазматических мембранах клеток-мишеней, но и оказывает влияние на АТ1-подобные рецепторы, расположенные на ядерной мембране; внутренней мембране МХ и лизосом. Участие валсартана в интракринной активности Ан II изменяет активацию генов и экспрессию их продуктов, контролирующих мышечное сокращение; перемещение из лизосом на ядро разнообразных регуляторов активности генов, включая экзогенный Ан II; регуляцию внутриклеточного Са2+, связанную с выходом Са2+ из Са2+-депо; образование радикалов кислорода, оксида азота и АТФ в МХ; активацию мегапоры МХ, индуцирующую апоптоз или некроз клеток.


