
Введение
Важнейшими медицинскими, социальными и экономическими проблемами современности стали ожирение и сахарный диабет 2 типа (СД2). По данным 2013 г., СД2 имели 374 млн человек. Избыток массы тела или ожирение в настоящее время имеют 526 млн. Во многом именно за счет данной когорты пациентов к 2030 г. на Земле будет 552 млн больных СД [1]. Стремительное распространение СД и ожирения в мире во многом обусловлено сложившимся за последние десятилетия рационом питания, где превалируют легкоусвояемые углеводы и животные жиры. Ранее считалось, что данная проблема коснулась в основном США, где был изобретен и массово применяется фаст-фуд. Однако, по данным 2012 г., в Российской Федерации 46,5 % мужчин и 51,7 % женщин имеют ожирение или избыток массы тела, а СД страдают 9–10 млн человек [2, 3]. Следовательно, проблема нерационального питания и гиподинамии, приводящих к развитию нарушений углеводного обмена, требует незамедлительных мероприятий здравоохранения. Изменение образа жизни на раннем этапе позволит избегать дальнейшего развития СД2, его осложнений и сердечно-сосудистой патологии.
Для решения данных проблем в развитых странах мира разработаны алгоритмы, основной целью которых является достижение гликемического контроля, однако такой односторонний подход недостаточно эффективен. В исследованиях ACCORD, ADVANCE и VADT строгий гликемический контроль сопровождался увеличением риска смертности. Согласно результатам NHANES III (1988–1994), 44,5 % пациентов с СД2 имели уровень гликированного гемоглобина (HbA1c) < 7 %, а в период наблюдения 1999–2000 гг. – только 35,8 % [4]. Следовательно, существуют дополнительные патогенетические аспекты СД2, открывающие перед врачами «новое окно возможностей». По современным данным, таким механизмом остается липотоксичность – комплекс негативных эффектов жирных кислот (ЖК) на органы и ткани, вовлеченных в патогенез СД. Связующее звено нарушений углеводного и жирового обменов – липотоксичность – лежит в основе новой патогенетической концепции СД2: Diabetus Mellipidus.
Липотоксичность-связующее звено нарушений углеводного и жирового обмена
В нескольких исследованиях показана взаимосвязь висцерального ожирения, резистентности к инсулину и гипергликемии. Висцеральное ожирение также является независимым сильным предиктором развития СД2. Висцеральный жир более метаболически активен, чем подкожный, в нем с высокой скоростью происходят процессы липолиза или липогенеза, что, соответственно, делает его основным источником ЖК. Висцеральная жировая ткань имеет ограниченные возможности хранения энергии в виде ЖК, индивидуальные для каждого человека. При избыточном длительном употреблении в пищу легкоусвояемых углеводов и жиров жировая ткань накапливает лишнюю энергию, однако, когда наступает предел ее возможностей, ЖК начинают поступать в кровоток и затем – в нежировые органы и ткани, не предназначенные для их хранения [5]. Насыщение адипоцитов ЖК также сопровождается активацией патологических механизмов: образуются более крупные, резистентные к действию инсулина адипоциты; запускается процесс местного воспаления, стимулируется секреция провоспалительных цитокинов, что способствует развитию нечувствительных жировых клеток. В поджелудочной железе, печени и мышцах запускаются различные патологические процессы, в комплексе способствующие развитию нарушений углеводного обмена.
При воздействии высокой концентрации свободных жирных кислот (СЖК) на культуру β-клеток и изолированные островки Лангерганса подавляется первая фаза глюкозостимулированной секреции инсулина [6]. По данным С. Olofsson и соавт., длительное воздействие на островки глюкозы и СЖК ингибирует секрецию инсулина на поздней стадии экзоцитоза, влияя на высвобождение инсулина при слиянии пор [7]. Кроме того, пальмитиновая кислота способна ингибировать экспрессию гена SUR (sulfonylurea receptor)-1, что также может являться одним из молекулярных механизмов подавления глюкозостимулированной секреции инсулина [8].
Помимо процессов высвобождения инсулина под действием СЖК нарушается экспрессия гена инсулина. В эксперименте на изолированных островках Лангерганса воздействие пальмитиновой кислоты вызывало увеличение концентрации церамида, что было ассоциировано со снижением количества мРНК инсулина. G. Solinas и соавт. показали, что пальмитиновая кислота активирует JNK в β-клетках, что сопровождается фосфорилированием субстратов рецептора инсулина 1 и 2, нарушая транскрипцию гена инсулина [9]. Более того, СЖК активируют апоптоз β-клеток путем образования церамида и активации оксидативного стресса. Синтез церамидов в β-клетках увеличивает количество адаптивных нитратоксид-синтаз. Результирующее увеличение оксида азота увеличивает экспрессию воспалительных цитокинов, включая интерлейкин-1 и фактор некроза опухоли, которые ухудшают функцию β-клеток и способствуют их апоптозу. Негативные эффекты СЖК на поджелудочную железу могут лежать в основе нарастающего снижения массы β-клеток и прогрессирования СД2.
Влияние СЖК на печень характеризуется несколькими механизмами.
В первую очередь происходит активация глюконеогенеза. Увеличение поступления СЖК в печень сопровождается ускорением окисления липидов и накоплением ацетил-КоА, повышенная концентрация которого стимулирует активность пируваткарбоксилазы и тормозит скорость ферментов глюконеогенеза, а также глюкозо-6-фосфатазы, контролирующей скорость высвобождения глюкозы из гепатоцитов. Кроме того, в исследовании G. Boden и соавт. показана способность СЖК подавлять ингибирующий эффект инсулина на гликогенолиз.
У пациентов с СД2 увеличение концентрации СЖК в плазме сопровождается повышением плазменного уровня глюкагона и чувствительности печени к активации гликогенолиза [10]. Дополнительным негативным эффектом СЖК в печени служит активация апоптоза гепатоцитов, что способствует развитию стеатогепатоза [11].
В мышечной ткани чрезмерное окисление ЖК приводит к накоплению внутриклеточного ацетил-КоА – мощного ингибитора пируватдегидрогеназы, торможения фосфофруктокиназы и накопления глюкозо-6-фосфата. Блокирование фосфорилирования глюкозы приводит к внутриклеточному накоплению глюкозы, сдерживающему транспорт в клетку через ГЛЮТ-4. Прямое действие длинноцепочечных жирных ацил-КоА на транспорт и фосфорилирование глюкозы также были продемонстрированы в мышцах: повышение их концентрации сопровождается ингибированием субстрата инсулинового рецептора 1 – фосфоинозитол-3-киназы – и ослаблением трансмембранного транспорта глюкозы [12]. Наконец, повышение церамида в мышцах (вторично к увеличению длинноцепочечных жирных ацил-КоА) нарушает транспорт глюкозы и подавляет гликогенсинтазу, активацию протеинкиназы В. В исследовании М. Bajaj и соавт. с участием пациентов с диабетом было показано улучшение чувствительности к инсулину в мышцах при применении аципимокса, снижающего уровень внутримышечного ацетил-КоА путем ингибирования СЖК [13]. Таким образом, повышение концентрации СЖК сопровождается развитием инсулинорезистентности в мышцах с помощью нескольких механизмов: ингибирования системы сигнализации инсулина, транспорта и фосфорилирования глюкозы, гликогенсинтазы, пируватдегидрогеназы.
Результаты клинических исследований подтверждают взаимосвязь липотоксичности и СД2. В исследовании М. Korani и соавт. общее содержание насыщенных и мононенасыщенных ЖК у пациентов с диабетом было значительно выше по сравнению с контролем (р = 0,006, р = 0,02 соответственно). Сывороточный уровень линолевой и полиненасыщенных ЖК у пациентов с диабетом были значительно ниже, чем в контрольной группе (р = 0,02). Заболеваемость диабетом значительно и положительно коррелировала с содержанием пальмитиновой, насыщенных и мононенасыщенных ЖК в плазме крови [14]. L. Wang и соавт. также показали взаимосвязь заболеваемости СД2 с уровнем ненасыщенных жирных кислот (пальмитиновой, стеариновой и пальмитолеиновой) [15]. Таким образом, в настоящее время имеются экспериментальные и клинические данные взаимосвязи СД2 и ожирения через механизм липотоксичности. В связи с неуклонной тенденцией СД2 к прогрессированию и развитию осложнений коррекция его патогенетических механизмов должна происходить с момента манифестации или даже на этапе начальных нарушений углеводного обмена.
Возможности ранней интервенции терапии с учетом патогенетических аспектов
На современном этапе эффективное управление СД2 возможно при многофакторном подходе, учитывающем все патогенетические аспекты заболевания (дисфункция β-клеток, инсулинорезистентность, глюкозо- и липотоксичность), ранней и индивидуализированной интервенции терапии, что позволит обеспечить долгосрочный метаболический контроль (рис. 1).
Эффективным и главное – патогенетическим методом коррекции патогенетических механизмов нарушений углеводного обмена является снижение массы тела и объемов висцерального жира. Это позволяет снижать уровень маркеров липотоксичности (т. к. ограничивается источник СЖК) и добиваться гликемического контроля, тем самым улучшая функцию β-клеток и чувствительность периферических тканей к инсулину. A.M. Rosenfalck и соавт. изучали долгосрочное влияние изменений «состава тела», вызванных снижением массы тела, на чувствительность к инсулину, инсулиннезависимое распределение глюкозы и функцию β-клеток поджелудочной железы. На фоне снижения массы тела и объема жира авторы зафиксировали статистически достоверное снижение уровня глюкозы натощак и нормализацию показателей постпрандиальной гликемии (ППГ), улучшение чувствительности к инсулину, рассчитанное по минимальной модели Бергмана. Необходимо отметить, что улучшение чувствительности к инсулину существенно коррелировало с уменьшением массы жира (r = -0,83, p = 0,0026) [16]. В исследовании D. Kelley и соавт. было показано снижение уровня HbA1c на 1,5 %, СЖК – на 174 ммоль/л при снижении массы тела на 12 кг (12 % от исходного) [17]. Кроме того, при снижении массы тела уменьшается риск развития СД2. В 3-летнем исследовании у 1067 пациентов было выявлено снижение риска развития СД2 на 90 % при снижении массы тела всего на 5–7 % [18]. Таким образом, снижение массы тела обладает патогенетическим действием в отношении предотвращения и прогрессирования СД, однако пищевые привычки не позволяют пациентам длительно и успешно соблюдать рекомендованную диетотерапию. Примерно у 90 % пациентов масса тела возвращается обратно, превышая первоначальные значения [19]. В данной ситуации представляется интересным возможности фармакологических препаратов для снижения массы тела и коррекции метаболических показателей у пациентов на раннем этапе развития СД2.
В настоящее время метформин служит препаратом первого выбора лечения СД2, его получают по крайней мере 120 млн человек по всему миру [20]. К основной функции метформина относится уменьшение производства глюкозы в печени – в основном путем ингибирования глюконеогенеза [21]. Несколько механизмов были предложены для объяснения этого ингибирующего действия на глюконеогенез в печени, включая изменения в активности ферментов [22], или уменьшение поглощения печенью субстратов глюконеогенеза [23]. Под его воздействием происходит стимуляция АМФ-активируемой протеинкиназы (АМФК), что приводит к увеличению экспрессии белка SHP (Small heterodimer partner), который в свою очередь подавляет экспрессию фосфоенолпируват-карбоксикиназы и глюкозо-6-фосфатазы, участвующих в глюконеогенезе [24]. Кроме того, препарат стимулирует субстрат инсулинового рецептора IRS (insulin-receptor substrate)-2, что вызывает увеличение поглощения глюкозы печенью через белок – переносчик ГЛЮТ-1, тем самым повышая синтез гликогена [25]. Метформин-индуцированное снижение печеночного содержания липидов ассоциировано с увеличением окисления ЖК и ингибированием липогенеза, что предположительно происходит благодаря AMФK-активации [26]. Действительно, AMФK координирует изменения в метаболизме липидов печени и таким образом регулирует разделение ЖК между окислительным и биосинтетическим путями. Так, AMФK-активация метформином индуцирует фосфорилирование и инактивацию ацетил-КоА-карбоксилазы, которая одновременно является основным предшественником для биосинтеза ЖК и мощным ингибитором их окисления в митохондриях [27]. Кроме того, AMФK подавляет экспрессию липогенных генов, таких как синтазы ЖК [28]. Недавнее исследование, проведенное Е. Kim и соавт., показало, что метформин вызывает AMФK-опосредованное торможение экспрессии стеароил-КоА-десатуразы, ограничивающей скорость действия ферментов биосинтеза мононенасыщенных ЖК из насыщенных ЖК [29]. В клинических исследованиях показана способность метформина снижать окисление СЖК на 10–30 %, усиливать их реэстерификацию и подавлять липолиз [30]. Например, использование метформина (500 мг) 21 пациентом с СД2 сопровождалось снижением концентрации СЖК на 17 %, а окисления жиров – на 25 % [31]. В двойном плацебо-контролируемом исследовании с участием 90 пациентов с СД2 показано снижение уровней глюкозы, HbA1c, СЖК и HOMA-IR (Homeostasis Model Assessment of Insulin Resistance) при применении метформина в дозе 1700 мг/сут на протяжении 6 недель [32]. Кроме того, уменьшение уровня СЖК положительно коррелировало с улучшением параметров гликемического контроля. Известным фактом является снижение массы тела на фоне приема метформина, вероятно, обусловленное замедлением всасывания в желудочно-кишечном тракте. По данным исследования DPP (Diabetes Prevention Program), снижение массы тела в течение 2 лет составило 2–3 % [31].
Таким образом, метформин обладает благоприятным эффектом на основные патогенетические механизмы СД2: снижает маркеры липотоксичности, улучшает чувствительность к инсулину, позволяет поддерживать гликемический контроль и опосредованно улучшает функцию β-клеток.
Сибутрамин является селективным ингибитором обратного захвата серотонина и норадреналина на уровне центральной нервной системы. Его основной фармакологический эффект состоит в ускорении наступления и пролонгации чувства насыщения, что приводит к уменьшению потребления пищи. Увеличивает расход энергии за счет стимуляции термогенеза путем опосредованной активации β3-адренорецепторов. Действует на обе стороны баланса энергии и способствует снижению массы тела. Эффекты сибутрамина у пациентов с СД2 изучались в нескольких исследованиях. Снижение массы тела составило от -1,8 до -8,5 кг в зависимости от вида протокола, исходной сахароснижающей терапии и продолжительности лечения. В исследовании женщин с СД2 и ожирением прием сибутрамина в течение 6 месяцев сопровождался снижением массы тела на 9,61 ± 1,7 кг, индекса массы тела (ИМТ) – на 3,92 ± 0,54 кг/м2, окружности талии – на 8,04 ± 3,47 см. Также наблюдалось снижение параметров гликемии: уровня глюкозы натощак на 124,8 ± 8,5, ППГ – на 102,2 ± 51,9 мг/дл, HbA1с – на 2,73 ± 0,01 % (р ≤ 0,001). Кроме того, достоверно снизились уровни общего холестерина, липопротеидов низкой плотности (ЛПНП), триглицеридов, диастолического давления [32]. По данным исследования STORM, было показано улучшение гликемического контроля, липидного профиля, снижение общей массы тела на 11,2 ± 6,3 кг, общей массы жировой ткани – на 18 % и уменьшение объемов висцерального жира – на 22 % на фоне приема сибутрамина в течение 6 месяцев [33]. Изучение влияния снижения массы тела на фоне сибутрамина на уровень СЖК проведено в ограниченном числе исследований. Х. Gao и соавт. показали достоверное снижение уровня СЖК натощак в зависимости от степени потери массы тела в группе сибутрамина, также улучшились показатели гликемии, липидного профиля и индекса HOMA-IR [34].
В России Редуксин (сибутрамин + целлюлоза микрокристаллическая) изучался в рамках наблюдательной программы ВЕСНА. В течение 6 месяцев было достигнуто снижение окружности талии на 10,4 %, уменьшение массы тела – на 14,3 %. Кроме того, наблюдалось улучшение показателей липидного обмена, увеличение содержания антиатерогенных липопротеидов высокой плотности на 14 %, снижение содержания триглицеридов на 14 %, а ЛПНП и общего холестерина – на 13 %. Таким образом, Редуксин эффективно снижает массу тела, положительно влияет на параметры углеводного и жирового обменов.
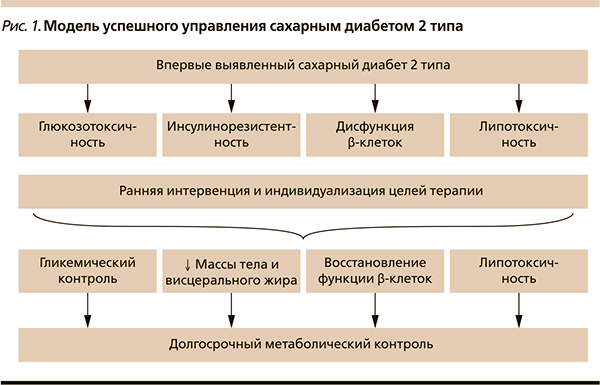
В настоящее время на кафедре эндокринологии и диабетологии РМАПО проводится исследование управления липо- и глюкозотоксичностью у пациентов с впервые выявленным СД2. Задача-ми нашего исследования стали оценка комбинированного влияния Редуксина (сибутрамин + целлюлоза микрокристаллическая) и метформина на маркеры липотоксичности, объем и/или перераспределение висцерального жира, определение динамики массы тела на фоне исследуемой терапии, оценка эффективности данной терапии на параметры гликемического контроля. В исследовании приняли участие 60 пациентов с длительностью СД2 не более 6 месяцев, продолжительность наблюдения – 24 недели. Всем пациентам рекомендовали диетотерапию с ограничением калорийности до 1500 ккал/сут. Они были распределены на основную группу, в которой 30 пациентов получали метформин и Редуксин, и группу сравнения, где применяли только бигуанид. Все участники соответствовали критериям включения и не имели противопоказаний к приему исследуемых препаратов. Группы пациентов были сравнимыми по возрасту, росту, антропометрическим и метаболическим характеристикам. Средний ИМТ составил 39,1 ± 12,8 кг/м2, также все пациенты имели висцеральное ожирение, по данным индекса объема талии/бедер – 0,97 ± 0,1. Для непосредственной оценки объема жировой ткани и висцерального жира использован метод двухэнергетической рентгеновской абсорбциометрии (DEXA). Средний объем жировой ткани в области туловища составил 24 кг, общая масса жировой ткани – 42,4 ± 3,72 кг. Среднее содержание жировой ткани составило 44,9 % (у здоровых людей оно не превышает 25 %), индекс мужского/женского жира в среднем – 1,16 ± 0,22 (в норме – 1) и положительно коррелировал с индексом объема талии/бедер (r – 0,61).
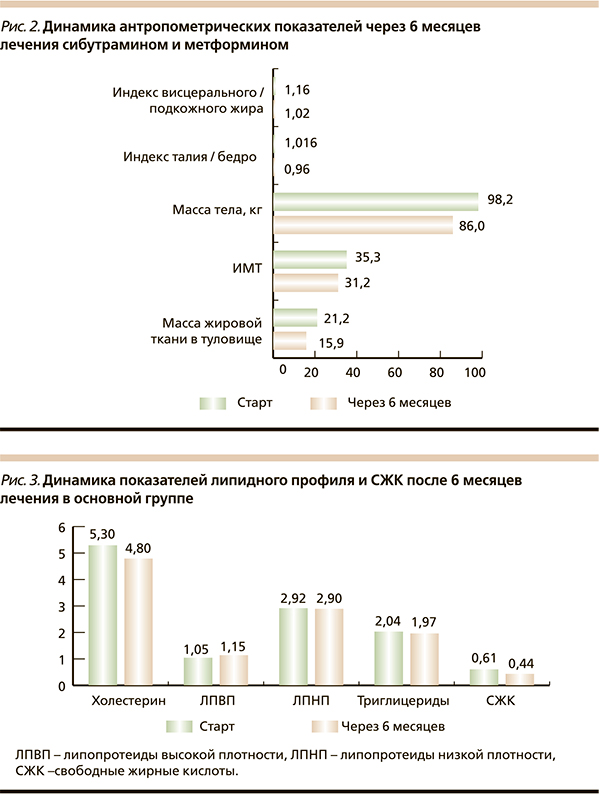
Общая масса жировой ткани положительно коррелировала с содержанием висцерального жира, систолическим артериальным давлением и уровнем общего холестерина. Таким образом, в абдоминальной области у пациентов превалировала висцеральная жировая ткань, метаболически наиболее неблагоприятная. Через 6 месяцев лечения в основной группе наблюдалось снижение массы тела с 98,1 ± 18 до 86 ± 15,6 кг, индекс объема талии/бедер уменьшился на 0,056 (p ≤ 0,05), индекс висцерального подкожного жира снизился на 0,14 (p ≤ 0,05), достоверных изменений данных характеристик в группе сравнения не выявлено (pис. 2). На фоне проводимой терапии метаболические параметры пациентов улучшились. Средний уровень СЖК в основной группе составил 0,61 ± 0,204 мгэкв/л (норма – 0,5), а после лечения – 0,44 ± 0,17 мгэкв/л. В основной группе уровень общего холестерина достоверно снизился с 5,3 ± 1,3 до 4,8 ± 1,01 ммоль/л (p ≤ 0,05) по сравнению с группой метформина. Уровень триглицеридов достоверно не изменился в группе лечения, а в группе сравнения их уровень увеличился (p ≤ 0,05). Данная зависимость была характерной также для ЛПНП (рис. 3). На старте исследования гликемический контроль был неудовлетворительным: средний уровень HbA1c составил 6,86 ± 0,85 %, гликемия натощак – 7,3 ± 0,91, постпрандиальная гликемия – 9,5 ± 1,31 ммоль/л. После терапии в основной группе уровень HbA1c достоверно снизился до 6,32 ± 0,74 % (p ≤ 0,05). Таким образом, у пациентов, получавших комбинированное лечение исследуемыми препаратами, наблюдалось эффективное снижение массы тела с уменьшением количества висцерального жира, улучшились показатели липидного профиля и гликемического контроля.
Параллельно развивающиеся неинфекционные эпидемии СД и ожирения имеют не только эпидемиологическую связь, но и общий патогенетический механизм. Воздействие на механизм липотоксичности в дебюте нарушений углеводного обмена может улучшить дальнейший прогноз и обеспечить метаболический контроль. В его основе лежит тенденция к накоплению ЖК в тканях и органах, что сопровождается нарушением в основных процессах углеводного и липидного обменов, а также активацией гибели β-клеток и гепатоцитов. В настоящее время воздействие на данный механизм возможно через снижение массы тела, объема висцерального жира и маркеров липотоксичности. Таким образом, своевременное и эффективное влияние на массу тела под контролем параметров гликемического и липидного профиля является многообещающим способом предотвратить или приостановить дальнейшее прогрессирование СД2.


