
Фармакогенетика изучает генетические особенности реакций организма на лекарственные препараты и способна помочь в индивидуальном подборе цитостатиков. Молекулярным субстратом служат вариации ДНК генов, кодирующих белки, которые отвечают за транспорт и метаболизм препаратов или служат их мишенями. В зависимости от распространенности в популяции эти варианты делят на
полиморфизмы (с частотой выше 1 %) и мутации (более редкие) [1–3].
Фармакогенетическое тестирование применяют для выявления генетических вариантов, ведущих к изменению активности белков, которые играют роль в фармакокинетике или фармакогенетике того или иного препарата. Теоретически фармакогенетическое тестирование может позволить скорректировать дозы или даже схему лечения, повысив его эффективность и снизив токсичность.
Ниже будут рассмотрены примеры фармакогенетического тестирования, которые уже вошли в клиническую практику (для индивидуализации химиотерапии иринотеканом, фторпиримидинами и тиопуринами), а также ряд других активно изучаемых клинико-генетических корреляций.
Глюкуронилтрансфераза и иринотекан
Глюкуронилтрансфераза соединяет с глюкуроновой кислотой ряд эндогенных (прежде всего билирубин) и экзогенных субстратов. Существует по крайней мере 17 изоформ этого фермента, объединенных в 2 семейства – UGT1 и UGT2. Ген UGT1A кодирует 9 изоформ подсемейства UGT1, все
они образуются за счет альтернативного сплайсинга. Наибольшее значение имеет изоформа UGT1A1, она отвечает за глюкуронирование билирубина. В гене UGT1A описано более 60 полиморфизмов, многие из которых лежат в основе синдрома Жильбера (легкая форма наследственной непрямой
гипербилирубинемии).
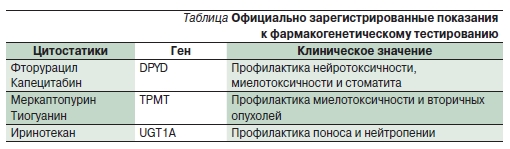
Рисунок 1. Метаболизм иринотекана СНЗА4 (цитохром Р450 3А4) превращает его в неактивные метаболиты NPC и АРС, СЕ (карбоксиэстераза) активирует с образованием SN-38, последний под действием UGT1А1 переходит в неактивный глюкуронид, бактериальная глюкуронидаза (d-Glu) в кишечнике катализирует обратную реакцию [4].
К субстратам UGT1A1А и UGT1A7 относится активный метаболит иринотекана – SN-38 (ингибитор ДНК-
топоизомеразы I, рис. 1). До 15 % населения являются гомозиготами по аллелю UGT1A1*2, сопряженному с увеличением от 6-го до 7-го числа повторов TA (тимин–аденин) в области промотора, что сопровождается снижением экспрессии гена и, соответственно, уменьшением количества белка.
В результате замедляется глюкуронирование SN-38, возрастает риск тяжелой нейтропении и поноса. На фоне высоких доз иринотекана (300–350 мг/м2) риск этих осложнений у гомозигот по аллелю UGT1A1*28 возрастал в 5–10 раз. Таким больным рекомендуется начинать лечение с дозы не выше 250 мг/м2, при хорошей переносимости она может быть повышена в последующих курсах. Для более низких доз корреляция между генотипом и побочными эффектами выражена слабо. Исследования последних лет указывают на более сложную картину: в ряде работ фармакокинетика и токсичность иринотекана лучше коррелировали с другими аллелями (например, UGT1A1*60 и UGT1A7*3); кроме того, по-видимому, имеют значение и другие гены, участвующие в метаболизме препарата и опосредующие его цитотоксичность [4, 5].
Дигидропиримидиндегидрогеназа и фторурацил
Дигидропиримидиндегидрогеназа участвует в катаболизме пиримидиновых оснований, а также инактивирует более 80 % фторпиримидинов (фторурацила и его производных – капецитабина, тегафура, флоксуридина). У 3–5 % европейцев активность фермента снижена, у 0,1–0,2 % он полностью отсутствует. До 50 % случаев недостаточности фермента связано с полиморфизмом IVS14+1G>A (аллель DPYD*2A): он заключается в замене одного нуклеотида, из-за чего нарушается сплайсинг мРНК, выпадает один экзон и образующийся укороченный белок быстро разрушается. Этот полиморфизм встречается у 1–2 % европейцев, с ним связывают около 25 % случаев тяжелой непере-
носимости фторурацила. Примерно так же распространен полиморфизм 2846A>T (т. е. замена аденина на тимин в 2846-м кодоне), вызывающий замену Asp949Val (т. е. аспарагиновой кислоты в 949-м положении на валин) и резкое снижение активности фермента [6, 7].
При выявлении этих полиморфизмов рекомендуется 2-кратное снижение дозы фторурацила и его аналогов или использование альтернативных схем, при носительстве двух вариантных аллелей эти препараты противопоказаны.
Тиопуринметилтрансфераза и меркаптопурин
Тиопуринметилтрансфераза катализирует S-метилирование тиопуринов (меркаптопурина, тиогуанина и азатиоприна), предотвращая образование активных метаболитов – тиогуаниновых нуклеотидов. Ген TPMT имеет полиморфные аллели, при некоторых стабильность фермента резко снижается. Описано 8 аллелей этого гена; наибольшее значение среди них имеют TPMT*2 (238G>C с заменой Ala80Pro),
TPMT*3A [460G>A (Ala154Thr) и 719G>A (Tyr240Cys)] и TPMT*3C (719G>A с заменой Tyr240Cys), причем у европейцев чаще встречается аллель TPMT*2, у афроамериканцев и азиатов –TPMT*3C. Примерно у 10 % людей активность фермента снижена, а у 0,3 % (гомозигот по указанным аллелям) практически отсутствует.
У детей с острым лимфолейкозом, получавших химиотерапию, недостаточность тиопуринметилтрансферазы вызывала угрожающее жизни угнетение кроветворения; из-за этого для
гомозигот по вариантным аллелям требовалось 10-кратное снижение доз, для гетерозигот – примерно 2-кратное. Возрастал также риск вторичных опухолей, в т. ч. глиом после профилактического облучения головного мозга. С другой стороны, наличие хотя бы одного из этих аллелей повышало
эффективность лечения [8].
P-гликопротеин
P-гликопротеин и его аналоги осуществляют АТФ-зависимый транспорт токсических веществ (включая цитостатики и их метаболиты) из клетки, а также участвуют в образовании гематоэнцефалического и других гистогематических барьеров [9].
Наибольшее значение для устойчивости к цитостатикам, включая антрациклины, таксаны, алкалоиды барвинка, иринотекан, топотекан, этопозид, ингибиторы тирозинкиназ, имеет P-гликопротеин (или белок MDR1), кодируемый геном ABCB1. Уровень экспрессии гена ABCB1 колеблется в широких пределах (у разных людей он может различаться в 50 раз), что обусловлено как генетическими, так и
средовыми факторами. На сегодняшний день описано более 50 аллелей гена ABCB1, наиболее распространены полиморфизмы в экзонах 12 (1236C>T), 21 (2677G>T/A) и 26 (3435C>T). Лишь
полиморфизм в 21-м экзоне влияет на аминокислотную последовательность, причем возможны 2 варианта: Ala893Ser и Ala893Thr. Основное внимание уделяется полиморфизму 3435C>T: хотя
он и не меняет структуру белка, в ряде экспериментов снижал стабильность мРНК, уменьшая количество P-гликопротеина. Несколько исследований показали более высокую токсичность и эффективность цитостатиков (антрациклинов, иринотекана, таксанов) у гомозигот по аллелю 3435Т
(ABCB1*6) или при наличии всех трех полиморфизмов (т. н. аллель P-gp*2). Впрочем, данные о функциональной и клинической значимости этих полиморфизмов неоднозначны, отчасти это может объясняться неравновесным сцеплением между полиморфными аллелями, т. е. вместе они встречаются
чаще, чем это объясняется их распространенностью в популяции [8, 9].
Заметный вклад в устойчивость к химиотерапии вносит белок – переносчик ABCG2 или BCRP (Breast Cancer Resistance Protein). Изначально описанный при раке молочной железы, он несколько отличается от P-гликопротеина по субстратной специфичности (переносит антрациклины, этопозид, иринотекан, топотекан, метотрексат, ингибиторы тирозинкиназ). Среди полиморфизмов, описанных для гена ABCG2, наиболее известны 421C>A (с заменой Gln141Lys и вероятным снижением активности) и 376C>T (с появлением стоп-кодона). Первый встречается среди 10 % европейцев и 30 % азиатов, второй выявлен лишь у азиатов [9].
Лекарственную устойчивость связывают также с аналогами P-гликопротеина из семейства MRP
(Multidrug Resistance-associated Protein), среди которых наибольшее значение имеют белкиереносчики MRP1 и MRP2 (их кодируют гены ABCC1 и 2). MRP2 находится на апикальной мембране эпителиальных
клеток, в частности гепатоцитов, и выводит в желчь различные эндогенные и экзогенные вещества в виде глюкуронидов и конъюгатов с глутатионом, в т. ч. билирубин. Белки семейства MRP имеют более узкую, чем P-гликопротеин, субстратную специфичность, но дополнительно обладают сродством к метотрексату и конъюгатам препаратов платины с глутатионом. Гены ABCC1 и ABCC2 также
имеют полиморфные аллели, вклад которых в эффективность и токсичность химиотерапии остается предметом изучения [9, 10].
Белки-переносчики семейства OATP
Важную роль в метаболизме лекарственных средств играют белкипереносчики семейства OATP (Organic
Anion Transporting Polypeptide), кодируемые генами SLCO или SLC21. Это семейство содержит 12 белков, среди которых три – OATP1B1, OATP1B3 OATP2B1 – находятся на базолатеральной мембраной гепатоцитов, обеспечивая захват печенью препаратов и естественных метаболитов [11, 12].
Белок OATP1B1 (кодируемый геном SLCO1B1) переносит билирубин, желчные кислоты, тиреоидные гормоны, эйкозаноиды, метотрексат, таксаны (паклитаксел и доцетаксел), лапатиниб, SN-38 (активный метаболит иринотекана), а также статины, цефоперазон, валсартан. Описано более 40 полиморфизмов гена SLCO1B1, ведущих к аминокислотным заменам.
Полиморфизм 521T>C встречается у 15–20 % европейцев и вызывает замену Val174Ala со снижением транспортной активности белка. Другой полиморфизм, 388A>G с заменой Asn130Asp, еще более распространен (до 40 % европейцев), но данные о его влиянии на активность белка неоднозначны.
В зависимости от сочетания генотипов возможны 4 варианта гена (аллеля): SLCO1B1*1A (388A + 521T), *1B (388G + 521T), *5 (388A + 521C) и *15 (388G + 521C). Распространенность этих аллелей в разных популяциях заметно отличается, чем, в частности, могут быть обусловлены этнические различия в фармакокинетике и фармакодинамике препаратов.
Белок-переносчик OATP1B3 обладает похожей структурой и по субстратной специфичности во многом
совпадает с OATP1B1 (из противоопухолевых препаратов переносит также иматиниб). Для гена SLCO1B3 описан ряд полиморфизмов с заменами аминокислот, в частности 334T>G и 699G>A, которые встречаются с частотой около 30 %.
Наличие вариантов SLCO1B1 или SLCO1B3, снижающих активность соответствующих белков-
переносчиков, способно замедлять печеночный метаболизм цитостатиков, увеличивая площадь под фармакокинетической кривой (AUC – Area Under the Curve) и усиливая токсичность. Показана связь фармакокинетики, токсичности и эффективности метотрексата с полиморфизмами гена SLCO1B1; аналогичные данные для таксанов пока несколько противоречивы [8, 11, 12].
Белки-переносчики семейств OСТ и MATE
Другая группа белков-переносчиков, вносящих существенный вклад в транспорт лекарственных средств, объединена в семейство OCT (Organic Cation Transporter). Белки OCT1, OCT2 и OCT3 (кодируются генами SLC22A1, 2 и 3) отвечают за попадание в клетки органических катионов. Белок OCT1 содержится главным образом в печени, OCT2 – в почках, OCT3 – в различных тканях [14].
Белок-переносчик OCT2 играет ключевую роль в фармакологии препаратов платины. Он находится на
базолатеральной мембране эпителия проксимальных извитых канальцев, обращенной к капиллярам, и захватывает различные органические катионы, действуя против градиента концентрации и используя мембранный потенциал. Затем эти вещества выводятся в мочу через апикальную мембрану белками-переносчиками семейства MATE (Multidrug And Toxin Extrusion, или SLC47A), MATE1 и MATE2-K. Субстратами описанной транспортной системы служат многие эндогенные соединения (креатинин, серотонин, триптофан, адреналин, дофамин, прогестерон), а также лекарственные средства (метформин, верапамил, β-адреноблокаторы, H2-блокаторы, ибупрофен, аспирин, амитриптилин, ламивудин), в т. ч. цитостатики – цисплатин, оксалиплатин, иматиниб и тамоксифен [13–15].
Цисплатин отличается лишь умеренным сродством к OCT2, еще более низким сродством к MATE1 и практически не взаимодействует с MATE2-K, из-за чего накапливается в клетках почечного эпителии, вызывая их гибель. Карбоплатин не взаимодействует с белками семейства OCT. Наконец, оксалиплатин лучше, чем цисплатин, захватывается белком OCT2, но при этом быстро выводится белками
MATE1 и MATE2-K, практически не накапливаясь в почечном эпителии.
Описан распространенный полиморфизм гена SLC22A2 808G>T, ведущий к замене аланина в 270-м положении на серин (Ala270Ser), снижающий сродство белка-переносчика OCT2 к ряду субстратов, включая цисплатин. У носителей этого полиморфизма отмечено снижение риска нефротоксичности [13, 15].
Цитохром P450 2D6
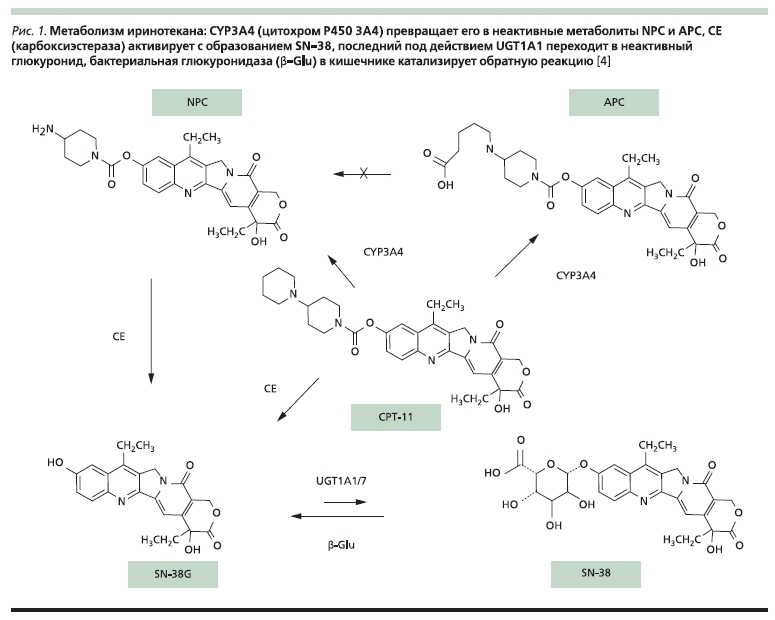
Субстратами CYP2D6 выступают до четверти лекарственных препаратов, среди противоопухолевых
средств наиболее важен тамоксифен (рис. 2). CYP2D6 катализирует реакцию окисления тамоксифена и
N-дезметилтамоксифена в 4-м положении с образованием, соответственно, 4-гидрокситамоксифена и эндоксифена, которые по антиэстрогенной активности превосходят тамоксифен в 50 раз. CYP3A4 и 3A5 наряду с другими изоферментами деметилируют тамоксифен и 4-гидрокситамоксифен [16].
Рисунок 2. Метаболизм тамоксифена [16].
Описано более 80 полиморфизмов гена CYP2D6, наиболее распространены и функционально значимы вариантные аллели CYP2D6*3 (2549delA со сдвигом рамки считывания), *4 (1846G>A с нарушением сплайсинга РНК), *5 (делеция всего гена), *6 (1707delT со сдвигом рамки), *10 (100C>T с заме-
ной Pro34Ser и снижением активности фермента), *41 (2988G>A с нарушением сплайсинга). Встречается также дупликация нормального аллеля CYP2D6*1 (может быть до 13 копий),
приводящая к повышению активности фермента. По сравнению с носителями двух нормальных аллелей при дупликации наблюдается примерно двукратное повышение уровня эндоксифена, а
при наличии двух нефункциональных аллелей – четырехкратное снижение. До 60 % европейцев имеют один нормальный и тот или иной вариантный аллельи но на активности фермента и результатах лечения это, по-видимому, сказывается незначительно. Однако 5–10 % людей не имеют ни одного
функционального аллеля, что чревато повышением риска рецидива рака молочной железы на фоне адъювантной терапии тамоксифеном [16–18]. В наиболее крупном исследовании Schroth и соавт., включившем 1325 больных с медианой времени наблюдения 9 лет, риск рецидива составил 14,9 % для носительниц двух нормальных аллелей CYP2D6, 20,9 % –для гетерозигот и 29 % – для гомозигот по вариантным аллелям; смертность была 16,7 %; 18,0 и 22,8 % соответственно [19]. Серия работ, в
которых изучали корреляцию генотипа с эффективностью тамоксифена в крупных рандомизированных испытаниях (включая ATAC и BIG 1-98), привела к неоднозначным результатам – во многом из-за неполного охвата больных, использования опухолевой ДНК вместо лейкоцитарной, а также генотипирования лишь по некоторым полиморфизмам. Кроме того, следует учитывать действие ингибиторов CYP2D6, в первую очередь антидепрессантов флуоксетина и пароксетина, которые
нередко прописывают таким больным [18].
Глутатион-S-трансферазы
Эти ферменты участвуют в инактивации препаратов платины, алкилирующих средств и антрациклинов. Наиболее изучены цитозольные изоферменты GSTA1, GSTM1, GSTP1 GSTT1. Для гена GSTA1 описано 3 распространенных полиморфизма в области промотора (-567T>G, -69C>T и -52G>A), снижающих экспрессию. До половины европейцев гомозиготны по делеции гена GSTM1, около 15 % – по делеции гена GSTT1. Наиболее частые полиморфизмы гена GSTP1 – 313A>G (замена Ile105Val) и 341C>T (Ala114Val). Аллель GSTP1 105Val достаточно распространен: его имеют около половины населения, причем 10–15 % из них составляют гомозиготы, аллель 114Val более редок (соответственно 15 % и 2–3 %). Изменение структуры фермента снижает его сродство к большинству субстратов (алкилирующим средствам и антрациклинам), но в отношении препаратов платины картина обратная:
в экспериментах in vitro инактивация этих цитостатиков ускорялась в 2–4 раза [20].
Аллель GSTP1 105Val улучшал результаты химиотерапии с использованием алкилирующих средств при раке молочной железы, лимфогранулематозе и миеломной болезни. В ряде работ аллель 105Val был сопряжен с улучшением прогноза больных раком толстой кишки, получавших оксалиплатин. Однако эти исследования носили ретроспективный характер и их результаты не нашли подтверждения в более крупных проспективных исследованиях. В других работах (при раке яичников и раке пищевода) аллель 105Val снижал выживаемость на фоне схем с цисплатином. Кроме того, у носителей аллеля 105Val, получавших цисплатин или оксалиплатин, отмечено снижение риска нейро- и ототоксичности. В целом накапливаются данные о повышении эффективности и усилении токсичности цитостатиков у больных
с вариантными аллелями глутатионS-трансфераз со сниженной активностью, хотя результаты и неоднозначны [21–23].
Белки репарации ДНК: ERCC1, XPD, XRCC1
Описано 2 полиморфизма гена ERCC1: 8092C>A (замена нуклеотида в 3′-нетранслируемой области гена) и 19007T>C (синонимичная замена Asn118Asn). Вариантные аллели не меняют аминокислотную последовательность белка, но могут влиять на экспрессию гена и, соответственно, устойчивость опухоли к цитостатикам. В эксперименте на клеточных линиях рака яичников аллель ERCC1 118T был сопряжен с уменьшенным количеством мРНК и трехкратным снижением способности к репарации
вызываемых цисплатином повреждений ДНК, хотя другая работа этого не подтвердила. Полиморфизмы
XPD 934G>A (Asp312Asn) и 2251A>C (Lys751Gln), а также XRCC1 1196G>A (Arg399Gln) влияют на строение и, по-видимому, активность соответствующих белков. Хотя можно было бы ожидать повышения эффективности химиотерапии у больных, имеющих вариантные аллели генов репарации ДНК, многие работы указывают на отсутствие статистически значимых корреляций или даже на обрат-
ную зависимость [22–26].
Заключение
На сегодняшний день фармакогенетическое тестирование одобрено FDA (американским фармкомитетом) в трех случаях – для фторпиримидинов, тиопуринов и иринотекана (см.таблицу); вероятно, этот список вскоре пополнит тамоксифен с CYP2D6.В некоторых клиниках фармакогенетическое тестирование вошло встандарты обследования перед назначением химиотерапии (особенно при детских лейкозах) и позволяет предотвращать многие случаи тяжелой
токсичности лечения.
Дальнейшее расширение этого списка потребует новых крупномасштабных экспериментальных и
клинических исследований, учитывающих как одновременный вклад нескольких белков в метаболизм
цитостатика, так и влияние более многочисленных, но сравнительно редких полиморфизмов обсуждавшихся в статье генов.



{kind=link}
{kind=link}