
Сахарный диабет (СД) 2 типа во всем мире признан одной из наиболее важных медико-социальных проблем современного здравоохранения вследствие постоянно растущей заболеваемости, высокой инвалидизации и смертности в работоспособном возрасте.
СД 2 типа относится к состояниям хронической гипергликемии, обусловленной прогрессирующей потерей чувствительности тканей к действию инсулина и относительной инсулиновой недостаточностью, ассоциированной с развитием макро- и микрососудистых осложнений. Клинически наиболее важна инсулинорезистентность (ИР) мышечной, жировой и печеночной тканей, которая приводит к многочисленным структуральным, гормональным и метаболическим последствиям. Поскольку 75 % глюкозы утилизируется скелетной мускулатурой, естественно, наиболее важным проявлением ИР может быть нарушение стимулированного инсулином захвата глюкозы скелетными мышцами. ИР мышечной ткани является наиболее ранним, возможно генетически определяемым, дефектом, проявляющимся снижением транспорта глюкозы и глюконеогенеза в миоцитах, намного опережающим клиническую манифестацию СД 2 типа.
На уровне жировой ткани ИР проявляется снижением антилиполитического действия инсулина. Это приводит к неконтролируемому окислению липидов и высвобождению большого количества свободных жирных кислот (СЖК), повышение уровня которых ведет к ингибированию процессов транспорта и фосфорилирования глюкозы и как следствие – к ослаблению окисления глюкозы, а также синтеза гликогена в мышцах. Избыток СЖК активизирует процессы глюконеогенеза, влияет на синтез липопротеидов в печени, приводя к повышенному образованию липопротеидов очень низкой плотности (ЛПОНП) и триглицеридов (ТГ), что сопровождается снижением уровня липопротеидов высокой плотности (ЛПВП). Кроме того, постоянное повышение уровня СЖК в плазме способствует аккумуляции липидов в островковых клетках поджелудочной железы, оказывая липотоксичный эффект на β-клетки, нарушающий их функцию.
ИР гепатоцитов характеризуется снижением синтеза гликогена и активацией процессов гликогенолиза, а также повышением продукции глюкозы de novo из аминокислот, лактата, пирувата, глицерина (глюконеогенез), в результате чего ее поступление в кровоток повышается.
Компенсаторным ответом на ИР независимо от ее причины является увеличение секреции инсулина в β-клетках поджелудочной железы и его избыточное выделение в кровь. В результате относительной или абсолютной гиперинсулинемии уровень глюкозы крови поддерживается в пределах нормального диапазона. Впоследствии длительная ИР, избыточные алиментарные потоки углеводов и жиров, хронически повышенная секреторная активность β-клеток, “липотоксичность” приводят к постепенному истощению секреторной способности β-клеток, клинической манифестации углеводных нарушений и дебюту СД. Гипергликемия замыкает “порочный круг”, вызывая снижение чувствительности β-клеток к действию глюкозы и других стимулов, что усиливает выраженность секреторного дефекта β-клеток (“глюкозотоксичность”). Сформулированная выше концепция развития СД 2 типа (см. рисунок) основана на наличии двух фундаментальных патофизиологических дефектов: ИР и дисфункции β-клеток поджелудочной железы, причем оба феномена существуют, взаимообусловливая и отягощая друг друга, и присутствуют в течение многих лет до манифестации заболевания.

Изучение ИР показало, что это чрезвычайно вариабельный параметр. При его определении с помощью прямого кламп-метода опосредованная инсулином утилизация глюкозы, даже у лиц с нормальной толерантностью к ней, может колебаться в значительных пределах, приближаясь в 25 % случаев к характерной для СД 2 типа. Первоначально нормогликемия у таких пациентов поддерживается за счет компенсаторной гиперсекреции инсулина, а гипергликемия развивается лишь при истощении β-клеток поджелудочной железы. При развитии СД 2 типа имеющаяся ИР прогрессирует. При определении ее степени с помощью того же кламп-метода выявляются достоверные различия в снижении скорости утилизации глюкозы у лиц с нормальной толерантностью к глюкозе и больных СД 2 типа. Причем отмечается обратная зависимость скорости утилизации глюкозы от уровня гликемии натощак: чем выше концентрация глюкозы крови, тем меньше скорость ее поглощения тканями.
В настоящее время ИР продолжают рассматривать как важнейшее патофизиологическое нарушение, присущее СД 2 типа, предшествующее декомпенсации функциональной активности островковых клеток поджелудочной железы и, возможно, ответственное за формирование сердечно-сосудистых нарушений, а также онкологическую предрасположенность. Собственно ИР – это нарушение биологического ответа клеток инсулинчувствительных тканей на воздействие эндогенного или экзогенного инсулина при достаточной его концентрации в крови. Свое биологическое действие на уровне клетки инсулин осуществляет посредством связи со специфическим инсулиновым рецептором, который представляет собой белок, состоящий из двух α- и двух β-субъединиц. Альфа-субъединица располагается на наружной поверхности клеточной мембраны и обеспечивает связь с инсулином, а трансмембранно расположенная β-субъединица обладает тирозинкиназной активностью. При связывании инсулина с α-субъединицей рецептора его β-субъединица активируется – проявляется способность тирозинкиназы фосфорилировать внутриклеточные субстраты инсулинового рецептора (СИР). В настоящее время хорошо изучены СИР-1, СИР-2, а также белки семейства STAT (signal transducer and activator of transcription). Фосфорилирование СИР ведет к плейотропной реакции клетки на действие инсулина. При посредничестве СИР-1 инсулин активирует один из ведущих ферментов, участвующих в передаче инсулинового сигнала, – фосфатидилинозитол-3-киназу, что приводит к стимуляции транслокации глюкозных транспортеров, в первую очередь GLUT-4, обеспечивающего трансмембранный перенос глюкозы в мышечные и жировые клетки. Ингибирование фосфатидилинозитол-3-киназы подавляет и базальное, и стимулированное инсулином поглощение глюкозы, а следовательно, транслокацию GLUT-4 к мембране (M. Reaven Gerald, 1999).
Изучение генетических факторов, обусловливающих развитие ИР, показало ее полигенный характер. В ее формировании могут иметь значение различные мутации генов СИР-1, гликогенсинтетазы, гормончувствительной липазы, β3-адренорецепторов, фактора некроза опухоли α, а также молекулярные дефекты белков, передающих сигналы инсулина, в частности увеличение экспрессии СИР-1 ингибитора тирозинкиназы инсулинового рецептора в мышечной ткани плюс снижение концентрации и активности внутриклеточных транспортеров глюкозы, например GLUT-4. Биологические эффекты инсулина заключаются в регуляции метаболических реакций (обмен углеводов, жиров и белков) и митогенных процессов (рост и дифференцировка тканей, синтез ДНК, транскрипция генов). Таким образом, понятие ИР не только касается метаболизма глюкозы, но и включает нарушение обмена белков, жиров, функции эндотелия и экспрессии генов.
Можно выделить ряд заболеваний и физиологических состояний, сопровождающихся ИР:
• физиологическая ИР (пубертатный возраст, беременность, диета, богатая жирами, ночной сон);
• метаболическая ИР (СД 2 типа, ожирение, декомпенсация СД 1 типа, избыточный прием алкоголя);
• эндокринная ИР (тиреотоксикоз, гипотиреоз, синдром Кушинга, акромегалия, феохромоцитома);
• неэндокринная ИР (эссенциальная артериальная гипертензия, цирроз печени, ревматоидный артрит, травма, ожоги, сепсис, хирургические вмешательства).
Механизмы, лежащие в основе ИР, окончательно не установлены. Так, результаты изучения процессов взаимодействия инсулина с клетками-мишенями позволяют выделить три группы механизмов, ответственных за развитие ИР: дорецепторный, рецепторный и пострецепторный. ИР, развивающаяся на дорецепторном уровне, обусловлена мутациями кодирующего гена. ИР на рецепторном уровне является следствием уменьшения числа рецепторов на поверхности клетки либо снижением их сродства к инсулину. Данные изменения могут быть генетически обусловленными или развиваться под влиянием внешних факторов. Но в большинстве случаев ИР вызвана дефектами передачи инсулинового сигнала на пострецепторном уровне, которые обусловлены структурно-функциональными нарушениями со стороны белков, вовлеченных в детерминацию сигнальных процессов. Однако молекулярные механизмы, определяющие развитие ИР, до сих пор интенсивно изучаются.
ИР и компенсаторная гиперинсулинемия играют ключевую роль в патогенезе нарушений углеводного обмена, что подтверждают данные многочисленных клинических и экспериментальных работ, однако они имеют многогранный потенциал для развития каскада других метаболических и сосудистых нарушений. Например, взаимосвязь ИР и гиперинсулинемии с нарушениями липидного обмена давно и широко обсуждается в литературе. По данным 3-го Национального исследования здоровья и питания (США) у 69 % пациентов с СД 2 типа имеет место дислипидемия. Крупное эпидемиологическое исследование San Antonio Study одним из первых продемонстрировало основные черты, характерные именно для диабетической дислипидемии: гипертриглицеридемию и снижение уровня ЛПВП. Причем была доказана достоверная корреляционная взаимосвязь между уровнем инсулина в крови, степенью периферической ИР и уровнем ТГ.
Многочисленными последующими исследованиями было подтверждено, что для лиц с ожирением и СД 2 типа наиболее характерно увеличение уровня ТГ и снижение концентрации холестерина (ХС) ЛПВП. Гипертриглицеридемия возникает в результате повышения продукции ЛПОНП, а также нарушения их катаболизма в печени. Проводимые ранее эпидемиологические и клинические исследования также показали, что увеличенный уровень ТГ является независимым фактором риска ИБС наряду с гиперхолестеринемией. Согласно сведениям, полученным в WHO Multinational Study, уровень ТГ у больных СД 2 типа имеет бoльшую прогностическую значимость в отношении возникновения ИБС, чем уровень общего ХС. Концентрация общего ХС крови и ХС липопротеидов низкой плотности (ЛПНП), как правило, находится на верхней границе нормы, однако уровень апо-В липопротеида – основного структурного и функционального белка ЛПНП, может быть увеличен. Причем наиболее выраженной является корреляция между высокой концентрацией ТГ, низкой концентрацией ХС ЛПВП и важнейшим маркером ИР – абдоминальным ожирением. Связь между абдоминальным распределением жира и уровнями общего ХС и ХС ЛПНП более сомнительна и значительно варьируется, по данным различных источников. Однако при оценке качественной характеристики ЛПНП у больных СД 2 типа и абдоминальным ожирением было продемонстрировано значительное повышение продукции малых плотных частиц ЛПНП, что делает их более атерогенными, даже в отсутствие количественных изменений. Изменение в структуре липопротеидов возникает вследствие перекисного окисления липидов и неферментативного окисления аполипопротеидов, входящих в их составе. Таким образом, сочетание гипертриглицеридемии, низкого уровня ХС ЛПВП и преобладание в крови малых плотных ЛПНП при близком к нормальному значению ХС ЛПНП характеризуют диабетическую дислипидемию, которая приводит к раннему развитию коронарного атеросклероза.
Помимо влияния ИР на развитие и прогрессирование дислипидемии при СД 2 типа нарушения липидного обмена в свою очередь воздействуют на выраженность ИР. На уровне транспорта глюкозы при ожирении и дислипидемии снижается количество транспортеров глюкозы в жировой и мышечной ткани. Нарушается также утилизация глюкозы на периферии, в первую очередь за счет уменьшения активности гликогенсинтетазы. Предполагается наличие генетического дефекта данного фермента при СД 2 типа, т. к. уменьшение массы тела пациентов с ожирением и СД 2 типа улучшает, но не устраняет данный дефицит. Влияние липидных нарушений на ингибирование гликогенсинтетазы нашло подтверждение в ряде работ: при росте содержания и метаболизма СЖК на фоне абдоминального ожирения образуется избыток ацетил-кофермента А (ацетил-КоА), который тормозит утилизацию глюкозы в гликоген даже без наличия генетической предрасположенности. Избыток ацетил-КоА также оказывает повреждающее действие на β-клетки поджелудочной железы у лиц с ожирением. Следовательно, можно говорить о взаимном влиянии липидного, углеводного метаболизма и феномена ИР. Нарушения липидого обмена при ожирении, особенно абдоминального типа, и СД 2 типа могут вызывать и/или усугублять ИР, а последняя может быть причиной возникновения и прогрессирования дислипидемий.
С учетом лежащего в основе СД 2 типа двойного метаболического дефекта (ИР и нарушение секреции инсулина β-клетками поджелудочной железы) стратегия и тактика управления данным заболеванием должны быть патогенетически обоснованы и направлены на устранение обоих нарушений. С этих позиций наиболее рациональным становится акцент на раннюю коррекцию ИР как наиболее важного дефекта, что позволяет достигать эффективной коррекции метаболических и гемодинамических нарушений, а также профилактики микрососудистых осложнений и сердечно-сосудистых заболеваний. Как показали результаты исследования UKPDS, у пациентов, получавших метформин, достоверно реже (на 41 и 39 % соответственно) развивались инсульты и инфаркты, а смертность снижалась на 42 %. Надо отметить, что и через 10 лет после завершения UKPDS эти пациенты оставались более благополучными в отношении всех указанных событий, а также других диабетических микрососудистых осложнений независимо от стабильности компенсации СД и уровня гликированного гемоглобина. Согласно консенсусному алгоритму управления СД 2 типа, предложенному Американской и Европейской диабетическими ассоциациями, рекомендовано раннее назначение метформина уже на этапе установления диагноза. Таким образом, с 2006 г. метформин (Формин Плива) в эффективной дозировке до 2000 мг/сут стал базовым средством для лечения СД 2 типа, а также лучшим компонентом для комбинированной терапии. Метформин должен назначаться независимо от возраста и веса на этапе установления диагноза СД 2 типа; исключением могут быть только пациенты с непереносимостью препарата или противопоказаниями к его приему в связи с каким-либо сопутствующим заболеванием.
Основными противопоказаниями к назначению метформина являются:
• нарушение функции почек (креатинин более 132 мкмоль/л для мужчин и более 123 мкмоль/л для женщин, скорость клубочковой фильтрации менее 60 мл/мин);
• лактоацидоз и указания на него в анамнезе;
• заболевания острые и хронические, которые сопровождаются гипоксиией (сердечная или дыхательная недостаточность, ХОБЛ, острый инфаркт миокарда, анемия);
• регулярный прием алкоголя.
Известны следующие фармакодинамические эффекты метформина, имеющие отношение к лечению СД 2 типа:
• усиление транспорта глюкозы в гепатоцит, усиление глюконеногенеза;
• уменьшение выраженности ИР и компенсаторной гиперинсулинемии;
• усиление утилизации глюкозы в периферических тканях;
• увеличение числа рецепторов к инсулину на клеточных мембранах и активизация транспортеров глюкозы в тканях-мишенях;
• уменьшение всасывания углеводов и белков в желудочно-кишечном тракте;
• снижение синтеза СЖК и ЛПОНП, уменьшение уровней ХС ЛПНП и ТГ в крови;
• снижение массы висцеральной жировой ткани, замедление липолиза адипоцитов;
• повышение уровней глюкагоноподобного пептида-1;
• уменьшение процессов клеточной пролиферации.
Многие эффекты метформина реализуется за счет увеличения активности тирозинкиназы, гликогенсинтетазы, стимулирующих синтез фосфатидилинозитол-3-киназы, – ферментов, ответственных за утилизацию глюкозы и уменьшения гиперсимпатикотонии. Преимуществом метформина является влияние на уровень гликемии не только натощак, но и постпрандиально без риска развития гипогликемических состояний и благоприятное воздействие на массу тела, что обеспечивает высокий уровень безопасности гипогликемизирующей терапии.
На этапе комбинированного лечения консенсусный документ по управлению СД 2 типа рекомендует более широкое использование новых препаратов – инкретиномиметиков и тиазолидиндионов (ТЗД; глитазонов, инсулинсенситайзеров), наравне с такими известными лекарственными средствами, как инсулин и препараты группы сульфонилмочевины. Так, в частности, ТЗД являются лекарственными средствами, улучшающими чувствительность тканей-мишеней к действию инсулина, принципиально отличающимися от метформина механизмом. Единственным представителем этого важного класса противодиабетических средств, разрешенным к применению в Европе, на сегодняшний день остается пиоглитазон. ТЗД являются синтетическими лигандами ядерных γ-рецепторов, активируемых пролифератором пероксисом (peroxisome proliferator-activated receptor; PPARγ), обладающих высокой липофильностью и способностью проникать через клеточные мембраны; их относят к семейству транскрипционных факторов.
PPARγ преимущественно экспрессируется в адипоцитах и моноцитах, меньше – в скелетных мышцах, печени и почках; соответственно синтетические агонисты PPARγ (глитазоны) контролируют экспрессию генов, регулирующих углеводный и липидный обмен в мышцах и жировой ткани. Особое значение имеет регулирующее влияние PPARγ на дифференцировку преадипоцитов в относительно мелкие и зрелые адипоциты, которые лучше утилизируют глюкозу и накапливают СЖК, что способствует снижению их уровня в системном и портальном кровотоке, а также уменьшению глюконеогенеза в печени. Зрелые адипоциты обладают большим количеством инсулиновых рецепторов и транспортеров глюкозы, характеризуясь меньшей липолитической активность. Это благоприятно отражается на липидном обмене и процессах атерогенеза. Доказано повышение уровня ЛПВП на фоне приема пиоглитазона, уменьшение уровней ТГ и ЛПНП с изменением состава последних в сторону преобладания более крупных, менее атерогенных молекул. Улучшение липидного профиля на фоне приема пиоглитазона проявляется в первую очередь увеличением уровня ХС ЛПВП. Улучшение утилизации глюкозы связано не только со снижением уровня СЖК в плазме крови, но и с прямым воздействием агонистов PPARγ на процессы передачи инсулинового сигнала. Активация PPARγ способствует экспрессии белка, транспортирующего СЖК, увеличению уровня белков транспортеров глюкозы, в первую очередь GLUT-4 и субстратов инсулиновых рецептов (СИР-1 и СИР-2), повышает экспрессию гена адипонектина. Кроме того, увеличение активности PPARγ обеспечивает их существенную роль в регуляции белков, разобщающих окислительное фосфорилирование, а также подавляет экспрессию гена лептина и ингибирует экспрессию в жировой ткани фактора некроза опухоли α, что сопровождается снижением ИР и улучшением секреции инсулина β-клетками.
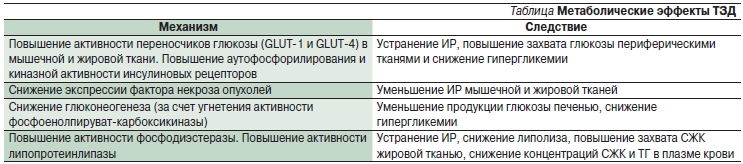
Пиоглитазон оказывают также протективное действие в отношении β-клеток, что может быть связано не только со снижением липотоксического действия СЖК, но и с непосредственным воздействием пиоглитазона на процессы апоптоза и пролиферации β-клеток. Усиление пролиферации β-клеток и снижение их гибели при применении ТЗД доказаны в экспериментальных работах. Таким образом, пиоглитазон (Амальвия) обладает комплексом положительных эффектов, связанных с углеводным метаболизмом, которые суммированы в таблице.
Известно, что на фармацевтическом рынке зарегистрировано только два препарата класса ТЗД (пиоглитазон и росиглитазон), которые имеют ряд негативных побочных эффектов, в частности вызывают задержку жидкости, способствуя развитию застойной сердечной недостаточности (СН), и увеличение массы тела. Были проанализированы результаты 7 исследований со средней длительностью 30 месяцев, в которых принимали участие 20 тыс. пациентов, подтвердивших увеличение риска развития застойной СН среди пациентов, получавших лечение ТЗД (относительный риск [ОР] – 1,72; 95 % доверительный интервал [ДИ] – 1,71–2,42; р = 0,002) в сравнении с контрольной группой. Несмотря на это, риск сердечно-сосудистой смертности на фоне лечения ТЗД не увеличился (ОР – 0,93; 95% ДИ – 0,67–1,29; р = 0,68). Не было также установлено различий в отношении риска сердечно-сосудистой смертности между росиглитазоном (ОР – 0,91; 95 % ДИ – 0,63–1,32; р = 0,63) и пиоглитазоном (ОР – 1,01; 95 % ДИ – 0,51–2,01; р = 0,98).
СН, возникшая на фоне лечения ТЗД, отличается от СН, развившейся вследствие систолической или диастолической дисфункции левого желудочка, и характеризуется более благоприятным прогнозом. Похоже, развитие застойной СН может быть следствием задержки жидкости, ассоциированной с увеличением давления в левом предсердии и легочного венозного давления, что демаскирует характерную для СД 2 типа бессимптомную диастолическую дисфункцию левого желудочка. Однако нет объяснений: почему СН, вызванная приемом ТЗД, и диастолическая СН имеют разные клинические исходы? По мнению авторов, развитие или усугубление имеющейся СН является характерным для этого класса препаратов эффектом, в связи с чем хроническая СН всех функциональных классов по NYHA является противопоказанием к назначению ТЗД пациентам с СД 2 типа.
Большое обсервационное исследование WellPoint Cohort Study проводилось с целью оценки риска развития инфаркта миокарда у пациентов, принимающих росиглитазон или пиоглитазон в реальной клинической практике. В него были включены 22 тыс. пациентов с СД 2 типа, принимающих росиглитазон, 23 тыс., получающих лечение пиоглитазоном, и 120 тыс. больных, принимающих другие пероральные сахароснижающие средства (ПССП).
Число случаев инфаркта миокарда составило 212 в группе росиглитазона, 232 – в группе пиоглитазона и 866 – на фоне терапии других ПССП. ОР инфаркта миокарда соответствовал 0,98 (95 % ДИ – 0,73–1,30) на фоне терапии росиглитазоном и 0,66 (95 % ДИ – 0,61–1,22) на фоне терапии пиоглитазоном в сравнении с терапией другими ПССП, оба показателя не достигли уровня статистической значимости. Исследование PROactive (Prospective Pioglitazone Clinical Trial In Macrovascular Events) продемонстрировало, что добавление 30–45 мг пиоглитазона к существующей терапии привело к снижению риска инфаркта миокарда на 28 % (p = 0,045). Напротив, несколько мета-анализов по росиглитазону показали значительное увеличение риска инфаркта миокарда и хронической сердечной недостаточности, хотя анализировались данные, собранные ретроспективно из нескольких краткосрочных исследований, не имевших цели оценить сердечно-сосудистые события.
На заседании Консультативного комитета FDA, который проходил 13–14 июля 2010 года, была представлена новая информация касательно безопасности росиглитазона и пиоглитазона для сердечно-сосудистой системы. Информация включала в себя данные по мета-анализам клинических испытаний росиглитазона и пиоглитазона, обсервационным исследованиям росиглитазона, включающим сравнение его с пиоглитазоном, и данные по росиглитазону, полученные на основании долгосрочных контролируемых клинических испытаний.
В целом мета-анализы и росиглитазона, и пиоглитазона не показали статистически значимого повышенного риска возникновения серьезных кардиальных событий. Однако в мета-анализах росиглитазона расчетный риск возникновения серьезных кардиальных событий при применении этого препарата был больше по сравнению с группами сравнения и почти статистически значимым. Наблюдался статистически значимый рост риска возникновения инфаркта миокарда и тяжелой ишемии миокарда. Данное явление не наблюдалось в мета-анализе пиоглитазона.
Таким образом, пиоглитазон (Амальвия) остается единственным эффективным и безопасным препаратом группы ТЗД, рекомендованным для медикаментозного лечения СД 2 типа, в основном на этапе комбинированной терапии. Однако необходимо помнить о потенциальных побочных эффектах, свойственных классу ТЗД, и назначать пиоглитазон с учетом известных противопоказаний.



{kind=link}