
Введение
Наследственные нарушения соединительной ткани (ННСТ) – спектр состояний, обусловленных генетически детерминированными аномалиями коллагена, фибриллина и других матриксных белков [1]. Клиническая картина обсуждаемых состояний неоднородна и значительно варьируется от минимальных особенностей развития опорно-двигательного аппарата, кожных, глазных или висцеральных проявлений до угрожающих жизни сердечно-сосудистых событий [2].
Особенностью недифференцированной дисплазии СТ (НДСТ) является отсутствие явного генетического дефекта с определенным типом наследования и, видимо, несколько меньшая степень риска сосудистых катастроф [3]. В действующих федеральных клинических рекомендациях по лечению пациентов с дисплазиями СТ среди медикаментозных средств ключевое место занимают препараты магния [4].
Магний – четвертый по содержанию в организме и второй по внутриклеточной концентрации катионом [3–5]. Ионы Mg2+ играют важнейшую роль в реализации таких клеточных функций, как интегрин-ассоциированная адгезия клеток на различных макромолекулярных субстратах, миграция клеток, транскрипция ДНК, синтез белка, обеспечивающих механический гомеостаз соединительной ткани [6]. Более глубокое понимание роли магния в патогенезе НДСТ и профилактике ее осложнений позволит персонифицировать походы к ведению лиц с обсуждаемой патологией.
Цель исследования: охарактеризовать современное понимание роли дефицита магния в патогенезе НДСТ.
Физиологическая роль магния в СТ
Магний участвует в поддержании нормального метаболизма и механического гомеостаза СТ.
Ионы Mg2+ стабилизируют вторичную и третичную структуры дезоксирибонуклеиновой и рибонуклеиновой кислот (ДНК и РНК), формируя катионные «мостики» между отрицательно заряженными фосфатными группами (рис. 1) [7]. Катион Mg2+ имеет решающее значение в поддержании конформации транспортной РНК, псевдоузлов РНК, элементов третичной структуры рибосомальной РНК, РНК-трансфер-мессенджера и каталитической самосплайсирующейся РНК (рис. 1) [7].
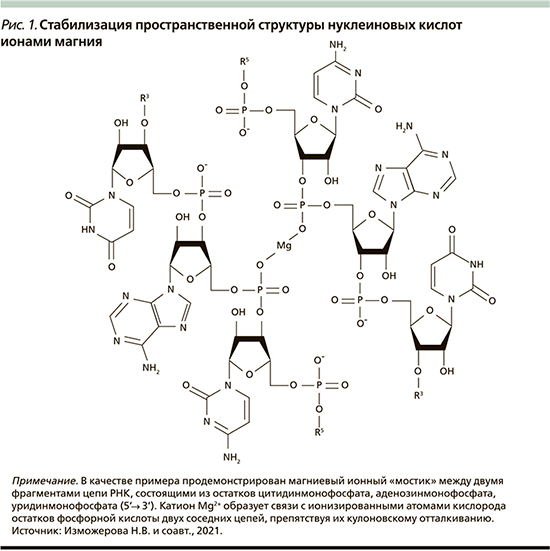
Магний оказывает влияние на фибриллярные компоненты (коллагеновые и эластические волокна) и нефибриллярные структурные элементы внеклеточного матрикса. Баланс между синтезом и деградацией этих структурных компонентов клетки определяет состояние цитоскелета клетки и его целостность.
Ремоделирование, деградация и протеолиз коллагеновых волокон внеклеточного матрикса обеспечиваются активностью матриксных металлопротеиназ (MMP) [8]. Эта группа металлоферментов имеет цинк-связывающий домен и включает коллагеназы-1, -2 и -3 (классифицируемые соответственно как ММР-1, -8, -13), расщепляющие фибриллярный коллаген типов I, II и III; желатиназы (ММР-2 и -9), разрушающие как коллаген-базальные мембраны, так и фибронектин; стромелизины (ММР-3, -10 и -11), действующие на различные компоненты внеклеточного матрикса, в т.ч. протеогликаны, ламинин, фибронектин и аморфные коллагены; а также семейство мембраносвязанных ММР ADAM (a disintegrin and metalloprotease), иначе именуемые адамазинами [9].
ММР продуцируются фиброблас-тами, макрофагами, нейтрофилами, синовиальными и некоторыми эпителиальными клетками. Секреция ММР индуцируется факторами роста (тромбоцитарный фактор роста [Platelet-derived growth factor – PDGF], факторы роста фибробластов [Fibroblast growth factor – FGF]), цитокинами (интерлейкин-1 (IL-1), фактором некроза опухоли (Tumor necrosis factor – TNF-α), фагоцитозом в макрофагах, а тормозится TGF-β и глюкортикоидами (рис. 2) [10].
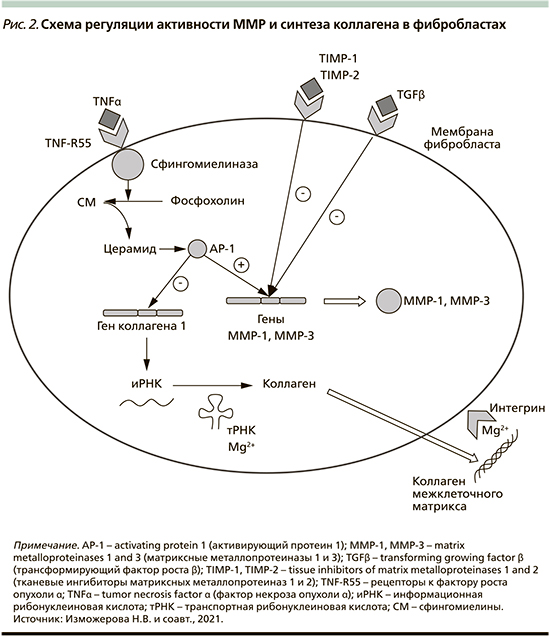
TNF-α также стимулирует деградацию внеклеточного матрикса, индуцируя экспрессию фибробластами MMP-1 и стромелизина-1 (MMP-3). Кроме того, ФНО-α ингибирует экспрессию гена коллагена I типа фибробластами, угнетает экспрессию генов эластина и декорина на транскрипционном уровне, способен блокировать активацию экспрессии генов коллагена и эластина I типа путем трансформации фактора роста β. Эффекты TNF-α на формирование внеклеточного матрикса частично перекрываются с эффектами интерлейкина-1 (IL-1), который также индуцирует экспрессию MMP-1 и -3 в фибробластах. Клеточные эффекты TNF-α опосредуются двумя различными рецепторами клеточной поверхности: TNF-RI (TNF-R55) и TNF-RII (TNF-R75), оба из которых экспрессируются фибробластами (рис. 2) [11].
Например, влияние TNF-α на экспрессию MMP-1, -3 и коллагена I типа в дермальных фибробластах в первую очередь опосредуется TNF-R55. Было показано, что связывание TNF-α с TNF-R55 активирует нейтральную сфингомиелиназу, ассоциированную с клеточной мембраной фосфолипазы, которая гидролизует структурный фосфолипид сфингомиелин клеточной мембраны до фосфохолина и церамида, липидного вторичного мессенджера [11]. Церамид стимулирует экспрессию генов MMP-1 и -3 посредством активации сигнальных путей, которые в конечном счете приводят к индукции AP-1 (activating protein-1)-зависимой транскрипции генов MMP (рис. 2). Кроме того, запуск церамидного пути в кератиноцитах человека приводит к сверхэкспрессии ММП-9 [9].
Эффект активации MMP зависит от наличия функционального AP-1-элемента в промоторной области гена MMP-1, а также от активности киназы, регулируемой внеклеточными сигналами 1/2 (ERK1/2), стресс-активируемой протеинкиназы/Jun N-концевой киназы (SAPK/JNK) и митоген-активируемой протеинкиназы р38 (MAPKs) [11].
Ингибирование матриксных металлопротеиназ происходит в основном за счет трансформирующего фактора роста β (TGF-β). TGF-β является важным фиброгенным стимулом, продуцируется большинством клеток грануляционной ткани, обусловливает миграцию и пролиферацию фибробластов, увеличение синтеза коллагена и фибронектина и снижение деградации внеклеточного матрикса путем ингибирования MMP [12].
Ионы Mg2+ необходимы для стабилизации некодирующих РНК.
В частности, ионы Mg2+ стабилизируют структуру транспортной РНК (тРНК) и дефицит магния приведет к увеличению числа дисфункциональных молекул тРНК, к снижению общей скорости белкового синтеза, хотя ионы Mg2+ непосредственно не взаимодействуют ни с молекулами коллагена, ни с TIMP [8].
Внутрисуставное введение сульфата магния кроликам в модели посттравматического остеоартрита снижало экспрессию информационной РНК (иРНК) интерлейкина-1β (IL-1β), TNF-α, MMP-3 в синовиальной оболочке [13]. Вместе с тем сульфат магния повышал продукцию мРНК основных компонентов внеклеточного матрикса: коллагена II типа и аггрекана – основного протеогликана, обеспечивающего гибкость, упругость и сжимаемость ткани [14]. Схожие результаты были получены при внутрисуставном введении хлорида магния, снижая экспрессию MMP-13 и интерлейкина-6 (IL-6) в синовиальной оболочке [15].
Использование препаратов магния в лечении пациентов с острым инфарктом миокарда тормозит повреждение миокарда. Концентрация IL-6 и ММП-1 в крови значительно возра-стает при остром инфаркте миокарда, но остается на сравнительно низком уровне у пациентов после терапии препаратами магния. Увеличение концентрации Mg2+ в сыворотке крови уменьшает уровень IL-6 и ММП-1 [16]. Насыщение диеты фолиевой кислотой и солями магния снижает секрецию ММП-2 и оказывает положительное влияние на течение ишемической болезни сердца [17].
Добавление магния уменьшало общую активность ММП-2 в клетках гладкой мускулатуры сосудов у крыс прямо пропорционально введенной дозе [18].
Патогенетическая роль дефицита магния в патологии СТ
Дефицит магния в ряде исследований отмечен как фактор риска развития сердечно-сосудистой патологии, что опосредованно как непрямым влиянием на липидный профиль, так и прямым повреждающим действием на сердечно-сосудистую систему [19]. Пролапс митрального клапана рассматривается как распространенное проявление дефицита магния [8, 20], а также синдрома Марфана – одного из вариантов ННСТ [21]. Дефицит магния ассоциирован с патологией митрального клапана [22].
Некоторые заболевания костной ткани, например остеопения или остеопороз, также могут быть ассоциированы с дефицитом магния [22, 23]. Тяжелый дефицит магния у матери может приводить к нарушению формирования костной ткани плода, что проявляется в виде несовершенного остеогенеза [24].
Дефицит магния также может приводить к гиперминерализации кости или аномального матрикса, в некоторых случаях с избыточным образованием остеоида [9, 10, 25–27], в результате могут развиваться т.н. мраморные, или меловые, хрупкие кости [24].
Дефицит магния в СТ приведет к замедлению синтеза всех структурных молекул и далее процессов восстановления, приводящих к ухудшению механических характеристик ткани [28].
Магний является важным фактором контроля пролиферации клеток за счет существенного влияния на синтез РНК, ДНК и белка. Митоз в значительной степени больше зависит от синтеза белка, чем от синтеза ДНК или РНК [7].
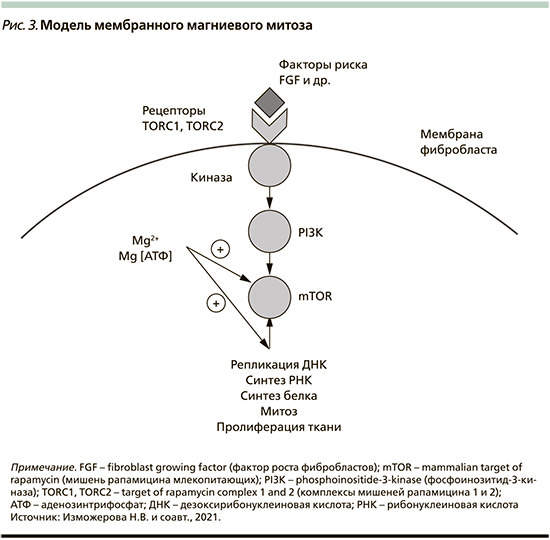
Исследования, идентифицирующие молекулярные медиаторы пролиферации Mg2+-зависимых клеток, привели к созданию модели мембранного магниевого митоза (МММ, рис. 3). Ключевым ее компонентом является белковый комплекс мишени рапамицина млекопитающих (mammalian target of rapamycin – mTOR), играющий главную роль в регуляции клеточного цикла. Согласно этой модели, факторы роста связываются с рецепторами сигнальных комплексов TORC1 и TORC2 (target of rapamycin complex 1 and 2) и вызывают фосфорилирование фосфоинозитид-3-киназы (PI3K), которая стимулирует комплекс mTOR. Активация mTOR зависит от внутриклеточного содержания комплекса MgАТФ, и АТФ был предложен в качестве основного регулятора активности mTOR. Однако при стимуляции клеток FGF изменяется концентрация не АТФ, а ионов Mg2+. Поэтому модель MMM предлагает Mg2+ в качестве основного регулятора динамики mTOR и пролиферации клеток [7]. Кроме того, повышенные уровни цитозольного Mg2+ способствуют рибосомной активности и синтезу белка, что в конечном итоге приводит к репликации ДНК и митозу [7].
Катион магния, связанный с эластическими волокнами, играет защитную роль в поддержании растяжимости элас-тина. С другой стороны, сообщалось, что магний увеличивает ферментативный гидролиз аортального эластина [28].
Устойчивый дефицит ионов магния приводит к синтезу неполноценного коллагена фибробластами. В частности, дефицит магния снижает активность Mg2+-зависимой аденилатциклазы, удаляющей дефектный коллаген. Это обуславливает беспорядочное расположение волокон коллагена и ведет к нарушению формирования СТ [29].
Отмечены структурные изменения коллагеновых и эластических волокон в стенке аорты у подопытных животных с дефицитом магния, связанные с экспрессией MMP-2 и -9 [30]. Сообщалось также о специфической локализации интегринов, функционирующих как трансмембранные рецепторы, и ММР, в связи с чем можно сделать предположение об их взаимосвязи и влиянии на функциональную активность ММР двувалентных катионозависимых конформационных изменений интегринов [31].
Также адгезия кератиноцитов и фибробластов к коллагену I типа и к гликопротеинам-ламининам базальной мембраны усиливалась магнием и снижалась катионами кальция [32]. Ионы Mg2+ участвуют в стабилизации структуры клеточных интегринов и в обеспечении их связывания с лигандами межклеточного матрикса (рис. 2) [33, 34]. При участии магния происходит перестройка низкоаффинной конформации интегрина α1β1 в высокоаффинную [34], повышая сродство последнего к коллагену и обеспечивая взаимодействие клетки с матриксом, крайне важное для пролиферации, дифференцировки и роста клеток [35]. Кроме того, взаимодействие Mg2+ с интегрином α2β1 играет важную роль в усилении пролиферации стромальных клеток посредством активации интегрин-сопряженной киназы фокальной адгезии (focal adhesion kinase, FAK) и связанных с ней сигнальных путей ERK (MAPK) и PI3K/Akt [36] (рис. 3).
Таким образом, магний принимает участие в реализации фундаментальных клеточных функций, таких как адгезия, миграция, синтез белка и пролиферация [34].
Антиоксидантная и антифибротическая активность литоспермата магния в терапии цирроза печени, индуцированного введением тиоацетамида, отмечена у крыс. Получено ингибирование фиброгенной активации звездчатых клеток печени [29], снижение уровня экспрессии эндотелием адгезионных молекул (ICAM1, VCAM1), подавление выработки провоспалительных цитокинов (TNF-α), торможение липополисахарид-индуцированной эндотелиальной дисфункции [37], а также повышение уровня оксида азота (NO), сосудистого фактора роста эндотелия (VEGF), эндотелиальной синтазы оксида азота (eNOS) и снижение системного артериального давления у крыс [38].
Метаболические нарушения при синдроме недифференцированной соединительнотканной дисплазии могут реализовываться в т.ч. и в сосудистой стенке, что обусловливает увеличение частоты встречаемости дисфункции эндотелия у лиц с НДСТ. Соединительнотканная дисплазия в данном случае является провоцирующим фактором вследствие повышения проницаемости эндотелия, фиброза сосудистых оболочек и прогрессирования структурной патологии артерий различного калибра [30]. Путем создания провоспалительной и протромботической среды в экспериментальных моделях установлено прямое влияние низкого содержания магния в организме на возникновение и прогрессирование эндотелиальной дисфункции. Установлено, что низкие концентрации магния обратимо ингибируют пролиферацию эндотелия и ингибирование пролиферации эндотелия осуществляется регуляцией синтеза интерлейкина-1 [22, 39].
Заключение
Магний выполняет разнообразные функции в СТ. Ионы Mg2+ стабилизируют нуклеиновые кислоты, участвуют в процессах синтеза белка и регуляции митотической активности фибробластов. Посредством различных механизмов магний подавляет избыточную активность матриксных ММР, препятствуя патологической деградации коллагеновых и эластических волокон. Кроме того, ионы Mg2+ регулируют конформацию клеточных интегринов, способствуя их адгезии на волокнах внеклеточного матрикса, обеспечивая структурную целостность СТ. Таким образом, магний играет ключевую роль в формировании и поддержании нормальной структуры СТ. Дефицит этого макроэлемента ассоциируется с нарушением функционирования фибробластов, дезорганизацией внеклеточного матрикса и развитием соединительнотканной дисплазии.
Вклад авторов. Изможерова Н.В., Шамбатов М.А., Попов А.А. – поиск источников литературы, обсуждение результатов, написание статьи. Бахтин В.М. – поиск источников литературы, обсуждение результатов, работа с иллюстрациями.


