
Введение
Прогероидные синдромы, или синдромы преждевременного старения, представляют собой клинически и генетически гетерогенную группу редких наследственных заболеваний, характеризующихся ускоренным старением организма. К прогериям и сегментарным прогероидным синдромам относят свыше десятка заболеваний, однако наиболее ярко признаки преждевременного старения проявляются при синдроме Хатчинсона–Гилфорда (прогерия детского возраста) и синдроме Вернера (СВ, прогерия взрослых) [1]. СВ впервые был описан O. Werner в 1904 г., а генетическая природа заболевания установлена и опубликована в 1996 г. C.E. Yu et al. Распространенность данного заболевания варьируется в различных странах по-разному, чаще всего синдром встречается в Японии (1/20–40 тыс.) и Сардинии (1/50 тыс.) [2]. В США, по данным NORD (National Organization of Rare Disorders), частота СВ составляет от 1 до 20 на 100 тыс.
В России нет регистра пациентов с СВ, имеются лишь отдельные его описания. В 2018 г. Е.Л. Соркина и соавт. описали трех пациентов с СВ [3]. Возраст манифестации синдрома варьируется от 15 до 30 лет. Медиана возраста диагностики составляет 37–40 лет [4]. Многие авторы отмечают, что одним из первых проявлений заболевания является отсутствие пубертатного ростового скачка с последующим развитием низкорослости. Маскообразное лицо, клювовидный нос и выступающий подбородок отличают больных СВ. На третьем десятилетии жизни отмечается появление седых волос, алопеция. Постепенное истончение кожи и атрофия подкожной жировой клетчатки приводят к развитию склеродермии [1].
Метаболические осложнения СВ включают развитие дислипидемии и инсулинорезистентности при нормальном показателе индекса массы тела (ИМТ) [5]. С течением заболевания формируются сахарный диабет 2 типа (СД2), требующий высоких доз инсулина, неалкогольная жировая болезнь печени (НАЖБП), гипогонадизм, остеопороз, атеросклероз, сердечно-сосудистая патология и онкологические заболевания. Риск развития злокачественных новообразований при СВ в 54 раза выше по сравнению с общей популяцией [6]. Чаще других описаны фолликулярная карцинома щитовидной железы, злокачественная меланома, менингеома, саркома мягких тканей, первичный рак костей и лейкемия/миелодисплазия [7, 6]. Двусторонняя катаракта, требующая оперативного вмешательства, описана у пациентов 20–30 лет, при этом для СВ характерно развитие задней субкапсулярной формы заболевания, а для пациентов с возрастными изменениями – нуклеарной формы [8, 9]. Для СВ типично развитие остеопороза, более выраженного в дистальных отделах конечностей, в отличие от стандартного возрастного снижения плотности костной ткани в позвоночнике [10, 11]. Из прочих нарушений отмечается метастатическая кальцификация мягких тканей. Чаще всего процесс локализован в ахилловом сухожилии, описаны случаи вскрытия кальцинозных очагов с формированием язв в месте процесса, вплоть до ампутации конечности [12]. По данным ряда исследователей, 30–40% пациенток с СВ способны к деторождению, однако к третьему десятилетию жизни происходит атрофия гонад с потерей фертильности [13, 14].
В большинстве случаев причиной летального исхода пациентов с СВ являются инфаркт миокарда и онкологические заболевания в возрасте до 50 лет [8, 10].
За развитие СВ ответственны мутации гена WRN, который находится на коротком плече 8-й хромосомы в локусе 8p12. Ген кодирует одноименный белок WRN размером 1432 аминокислоты, представляющий собой хеликазу семейства RECQ. Эти ферменты разделяют цепи двуцепочечной молекулы ДНК (ДНК-хеликазы) или внутримолекулярные связи в молекулах РНК (РНК-хеликазы), используя энергию гидролиза АТФ или ГТФ (гуанозинтрифосфат). Хеликазы движутся вдоль цепи нуклеиновой кислоты в направлении, характерном для каждого фермента. Мутации в гене WRN приводят к нарушению функции ДНК-хеликазы.
В результате возникают нарушения репликации и репарации ДНК, экспрессии генов, ускоренное укорочение теломер и повышенная чувствительность к апоптозу [15, 16]. На сегодня описано более 80 мутаций гена WRN; самые распространенные – миссенс-мутации (переключение кодона на кодирование другой аминокислоты – 65,28 %), несмысловые замены (замена нуклеотида в кодоне, не приводящая к изменению последовательности аминокислот в соответствующей полипептидной цепи, – 21,52 %) и нонсенс-мутации (в результате которых кодон теряет способность кодировать какую-либо аминокислоту и становится стоп-кодоном, что приводит к преждевременной терминации синтеза белка, – 5,87 %) [16]. Любая мутация независимо от того, затрагивает она хеликазную область или нет, приводит к полной потере белком своей функции. Вариабельность клинических проявлений заболевания, вероятно, связана со степенью экспрессии гена WRN в различных типах клеток и различным репликационным потенциалом клеток предшественников [10, 17].
Клинический случай
Обследование пациентки включило сбор анамнеза жизни, наследственность, отягощающие факторы, наличие сопутствующих заболеваний, анализ медицинской документации. Проведена динамическая оценка антропометрических показателей ребенка с использованием калькулятора AnthroPlus ВОЗ. Исследованы маркеры функции печени, липидограмма, глюкоза крови, оральный глюкозотолерантный тест. Проведена оценка уровня гормонов: тиреотропного гормона (ТТГ), свободного тироксина (Т4св), антител к тиреоглобулину (АТ-ТГ), антител к тиреоидной пероксидазе (АТ-ТПО), иммунореактивного инсулина (ИРИ), С-пептида, в т.ч. и на фоне лечения. Инструментальные методы диагностики включали ультразвуковое исследование (УЗИ) органов брюшной полости и малого таза, щитовидной железы (ЩЖ), электрокардиографию (ЭКГ). Молекулярно-генетическое исследование гена WRN у пробанда и членов семьи проводилось в лаборатории молекулярной генетики на базе отделения наследственных эндокринопатий ФГБУ ЭНЦ. После выписки из стационара ребенок наблюдался в течение года в условиях профильного эндокринологического центра.
Пробанд – девочка, впервые обследована в условиях эндокринологического стационара в возрасте 16 лет 11 месяцев. При поступлении предъявляла жалобы на задержку роста, низкие весовые прибавки, появление седых волос (с 10 лет жизни) и их потерю, ломкость ногтей (с 14 лет жизни). Из анамнеза известно, что девочка родилась в срок с нормальными показателями роста и массы тела. Национальность – азербайджанка. Ребенок от близкородственного брака, родители – кузены. Случаев анте- и перинатальной гибели детей в семье нет. Привита по национальному календарю прививок. Болеет нечасто. Травм, переломов, операций не было. Состоит на учете у ортопеда – комбинированный грудопоясничный сколиоз 2–3-й степеней, плоскостопие (установлены в 13 лет), кардиолога – нарушение ритма сердца (НРС), синусовая тахикардия (установлена в 13 лет), гастроэнтеролога – НАЖБП (установлена в 15 лет), невролога – головные боли напряжения, нарушение засыпания (с 16 лет жизни).
При осмотре выявлены низкорослость (-2,0 SDS), снижение массы тела (ИМТ=17,7, SDS ИМТ=-1,3). Отмечена сухость кожных покровов. Подкожно-жировой слой истончен в дистальных отделах конечностей. Сколиоз грудопоясничного отдела позвоночника. Отмечено: нос больших размеров клювовидной формы, диффузная алопеция и седина волос. Проведена динамическая оценка антропометрических показателей ребенка, данные представлены в таблице.
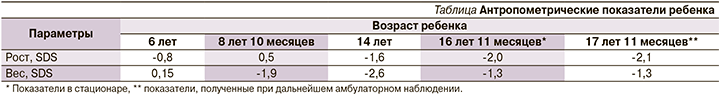
Как следует из таблицы, задержка роста сформирована к периоду пубертата: к 14 годам жизни -1,6 SDS роста, к 16 годам жизни низкорослость -2,0 SDS роста. Таким образом, у пациентки отмечено отсутствие пубертатного скачка роста с последующим развитием низкорослости. Дефицит массы тела сформирован в препубертатном периоде и в дальнейшем сохранялся: к 9 годам жизни -1,9 SDS ИМТ, к 16 годам 11 месяцам жизни -1,3 SDS ИМТ; при этом максимум снижения показателя отмечен в периоде пубертата (14 лет жизни ребенка) -2,6 SDS ИМТ. На момент госпитализации половое созревание у девочки соответствовало 5-й стадии по Таннер, присутствовали регулярные менструации.
Исследованы липидограмма, глюкоза крови, маркеры функции печени, проведена оценка показателей в динамике. На момент госпитализации все показатели липидограммы нарушены: повышены уровни холестерина крови (ХС) – 5,9 (3,0–5,2) ммоль/л, триглицеридов (ТГ) – 7,43 (0–2,3) ммоль/л, коэффициента атерогенности – 7,4 (≤3); снижен уровень липопротеидов высокой плотности (ЛПВП) – 0,7 (0,9–1,4) ммоль/л. Динамические показатели липидограммы представлены на рисунке.
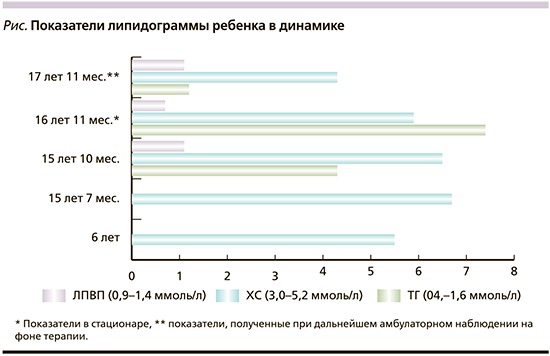
Как следует из рисунка, повышенный уровень ХС (5,5 ммоль/л) у девочки был выявлен в раннем возрасте (в 6 лет жизни) и длительно сохранялся (в 15 лет 10 месяцев, 6,5 ммоль/л). Отмечено нарастание уровня ТГ и снижение уровня ЛПВП с течением времени.
Проведена оценка показателей углеводного обмена (ИРИ, С-пептид, глюкоза крови). На момент госпитализации ребенка ИРИ составил 69,1 (2,6–24,9) мкЕД/мл, С-пептид – 9,9 (1,1–4,4) нг/мл. Проведенный оральный глюкозотолерантный тест не выявил нарушений углеводного обмена. Впервые ИРИ исследован в 14 лет жизни девочки, повышен до 36 мкЕД/мл.
В 14,6 года уровень ИРИ составил 138 мкЕД/мл, с целью коррекции инсулинорезистентности начата терапия метформином. Проведено УЗИ органов брюшной полости, подтверждена НАЖБП, при этом не отмечено значимого повышения традиционных сывороточных биомаркеров. Проведено УЗИ органов малого таза: размеры матки и гонад соответствовали 13 годам жизни ребенка.
Выполнено УЗИ ЩЖ, отмечены структурные нарушения (линейные гиперэхогенные включения). Проведена оценка функции ЩЖ, подтвержден субклинический гипотиреоз: ТТГ=6,09 (0,5–4,3) мкМЕ/мл, Т4св=13,7 (12,6–21,0) пмоль/л. Данных за аутоиммунный процесс в ЩЖ не получено, уровни АТ-ТГ и АТ-ТПО были в норме. Впервые повышение уровня ТТГ выявлено в возрасте 13 лет и подтверждено при динамическом наблюдении. Начата заместительная гормональная терапия L-тироксином, на момент госпитализации доза составила 25 мкг/сут.
Проведена ЭКГ, подтверждено НРС, синусовая тахикардия. Впервые НРС выявлено в возрасте 13 лет, комплексная оценка сердечно-сосудистой деятельности проведена в возрасте 16,8 года в условиях профильного консультативного центра, заподозрена пароксизмальная тахикардия.
Пациентка амбулаторно неоднократно консультирована окулистом, патологии не выявлено.
Проведено молекулярно-генетическое исследование семьи. У пробанда выявлена гомозиготная мутация с.3243_3244del:р.Т1081fs гена WRN (патогенная), гетерозиготный вариант с.392Т>С:р.V131А гена CAV1 (вариант неопределенной клинической значимости). У родной сестры и отца пробанда выявлены аналогичные гетерозиготные варианты в генах WRN и CAV1.
У матери пробанда выявлен аналогичный гетерозиготный вариант только в гене WRN. Родной брат девочки не является носителем данных изменений.
Терапия метформином начата в возрасте 14,6 года жизни, доза препарата последовательно увеличивалась и к моменту госпитализации составила 1700 мг/сут. В стационаре доза была увеличена до 2000 мг/сут. В стационаре начата терапия фенофибратом в дозе 145 мг/сут. Продолжено лечение L-тироксином, доза скорректирована до 37,5 мкг/сут.
Наблюдение ребенка проводилось до достижения возраста 18 лет. Низкорослость и снижение массы тела сохранялись (см. таблицу). Менструальный цикл у девушки был регулярным. В возрасте 17 лет 11 месяцев проведен лабораторный контроль: уровни гонадотропинов, эстрадиола, тестостерона соответствовали возрастным значениям, данных за гипогонадизм, синдром гиперандрогении не получено. Лечение продолжено в полном объеме. Несмотря на проводимую терапию метформином, инсулинорезистентность сохранялась. Показатели ИРИ и С-пептида оставались значимо высокими: ИРИ=137,5 мкЕД/мл, С-пептид=10,9 нг/мл. Проводился регулярный мониторинг гликемии, выявлена гипергликемия натощак до 7,4 ммоль/л. Отмечен положительный эффект приема фенофибрата: уровни ХС, ЛПВП, ТГ достигли физиологических значений (см. рисунок), коэффициент атерогенности снизился до 2,68. Субклинический гипотиреоз находился в стадии медикаментозной компенсации, ТТГ=1,5 мкМЕ/мл.
Обсуждение
По данным литературы, средний возраст диагностики заболевания составляет 37–40 лет [4]. Как правило, первые симптомы заболевания становятся очевидными при ретроспективном анализе данных пациентов с СВ. Согласно данным различных регистров, существует ряд фенотипических особенностей, характерных для СВ.
В нашем случае низкорослость с отсутствием пубертатного ростового скачка, изменение волос (седина, алопеция), заостренные черты лица, клювовидный нос, истончение подкожно-жирового слоя конечностей, дефицит массы тела у пациентки позволили заподозрить наследственную липодистрофию с прогероидными чертами уже при первичном осмотре. К моменту госпитализации у ребенка сформировались и прогрессировали метаболические нарушения (инсулинорезистентность, неассоциированная с набором массы тела, гиперхолестеринемия со снижением уровня ЛПВП, гипертриглицеридемия) и инструментально выявленные изменения (НАЖБП, НРС с развитием синусовой тахикардии). Совокупность фенотипических особенностей и лабораторных нарушений стала показанием к проведению генетических тестов. Достаточно ранняя диагностика заболевания в представленном случае, безусловно, связана с проведением молекулярно-генетического исследования. У девочки выявлен патогенный гомозиготный вариант в гене WRN. Дефект в том же кодоне гена WRN описан французскими учеными в составе биаллельных мутаций, обусловивших заболевание. В описанном клиническом случае представлен мужчина 48 лет с катарактой, остеопорозом и неопластическим процессом [18]. Также пациентка имеет дополнительную гетерозиготную мутацию в гене CAV1, ассоциированную с врожденной генерализованной липодистрофией 3-го типа и частичной липодистрофией с врожденной катарактой, нейродегенеративным синдромом (Cao et al., 2008). Однако отсутствие клинико-лабораторных изменений у родственников (отец и старшая сестра) с тем же вариантом позволяет предположить, что в данном случае гетерозиготное носительство не привело к каким-либо фенотипическим проявлениям.
На сегодняшний день специфического лечения СВ не существует. Модификация образа жизни и коррекция питания крайне важны для пациентов. Регулярные физические нагрузки приводят к улучшению метаболических показателей [1, 10]. Также необходимо придерживаться лечебного питания с ограничением легкоусвояемых углеводов и жиров (менее 30%/сут); в рационе должны присутствовать продукты с большим содержанием омега-3 ненасыщенных жирных кислот [1, 5]. Ведение пациентов с СВ основывается на симптоматической терапии с целью коррекции метаболических нарушений и предотвращения развития острых и хронических осложнений. Для коррекции липидных нарушений помимо диеты могут быть рекомендованы фибраты [19] и/или статины [4, 5]. В настоящей работе пациентка получала фенофибрат с хорошей переносимостью. Прием препарата привел к нормализации всех показателей липидного обмена. С течением времени у пациентов с СВ формируется СД2. Большинство препаратов, в частности, для лечения СД2 не разрешены к применению в отношении детей. Препаратом первой линии для коррекции СД2 являются метформин и/или инсулин.
На момент диагностики заболевания у пациентки, несмотря на длительное лечение метформином, сохраняется инсулинорезистентность, а к концу наблюдения отмечается гипергликемия натощак до 7,4 ммоль/л. Таким образом, несмотря на проводимую терапию, у ребенка продолжается формирование СД2. После достижения 18-летнего возраста для пациентов, сформировавших СД2, возможно применение препаратов ингибиторов дипептилпептидазы-4 и агонистов глюкагоноподобного пептида-1, тиазолидиндионов [20, 21]. При выраженных явлениях остеопороза рекомендовано назначение препарата группы бисфосфонатов в сочетании с витамином D3 и кальцием [11, 22].
Ведется активный поиск препаратов для потенциального лечения пациентов с СВ и другими прогероидными синдромами. Одно из наиперспективных направлений – исследования ингибиторов mTOR (сиролимус и его производные), используемых в настоящее время в составе иммуносупрессивной терапии [10, 23, 24]. Аналогичные исследования проводятся с ингибиторами MAPK (митоген-активируемые протеинкиназные каскады), участвующими в системе клеточного выживания (апоптотической активности) и пролиферации [25, 26]. Безусловно, вопросы медикаментозной терапии детей требуют дальнейшего изучения и наблюдения большего числа случаев.
Выводы
При сочетании таких фенотипических проявлений, как низкорослость, изменение волос (поредение, ранняя седина) и потеря подкожно-жировой клетчатки, целесообразно заподозрить наследственную липодистрофию, в частности, в структуре прогероидных синдромов.
Ранняя диагностика СВ позволит предотвратить острые состояния и осложнения заболеваний, ассоциированных с синдромом, а также сделает возможным использование репродуктивных технологий еще до наступления атрофии гонад.
Этиопатогенетическая терапия СВ не разработана, проводится симптоматическое лечение. Коррекция нарушений липидного обмена в детском возрасте фибратами и/или статинами может быть успешной. Препаратом первой линии для коррекции СД2 являются метформин и/или инсулин.
В семьях со случаями СВ обязательно проведение медико-генетического консультирования и психологической поддержки.
Благодарности. Авторы выражают благодарность сотрудникам «НМИЦ эндокринологии», участвовавшим в согласовании и проведении молекулярно-генетического исследования. Пациентке и семье пациентки.
Вклад авторов. Все авторы одобрили финальную версию статьи перед публикацией, выразили согласие нести ответственность за все аспекты работы, подразумевающую надлежащее изучение и решение вопросов, связанных с точностью или добросовестностью любой части работы.
Финансирование. Молекулярно-генетическое исследование выполнено в ФГБУ «НМИЦ эндокринологии» МЗ РФ, в рамках программы «Альфа-эндо».


