Агрессивный пародонтит (АП) – высокоактивный тяжелый воспалительно-деструктивный процесс, поражающий ткани пародонта, сопровождающийся потерей зубодесневого прикрепления, разрушением альвеолярной кости и образованием пародонтальных карманов [1].
В основе представлений обо всех воспалительных заболеваниях пародонта лежит микробная теория. Продолжительные исследования позволили выделить сначала микроорганизмы, а затем и их комплексы, специфичные для той или иной нозологии [2]. До сих пор ведутся дискуссии на предмет уточнения последовательности всех патогенетических звеньев развития АП.
Рост числа пациентов, их молодой возраст, «смазанные» начальные симптомы развития болезни, несвоевременная обращаемость к пародонтологу, тяжелые деструктивные процессы и как следствие – ранняя потеря зубов вынуждают искать новые способы ранней детекции предрасположенности к АП.
По мере накопления клинических и лабораторных данных о развитии, течении и патогенезе АП было выдвинуто предположение о наследственном механизме передачи этого заболевания, что косвенно подтверждается выявленной закономерностью возникновения и развития АП у членов одной семьи. Сейчас уже ни у кого не вызывает сомнений, что знание индивидуальных особенностей ДНК позволяет предсказывать вероятность развития тяжелых заболеваний и, что особенно важно, заблаговременно принимать меры по их предотвращению [5].
В основу концепции возникновения и развития как хронического, так и агрессивного пародонтита положена бактериальная биопленка. Установлено, что именно внедрение Aggregatibacter actinomycetemcomitans в ткани пародонта вызывает реакцию нейтрофилов с последующим выбросом медиаторов воспаления и цитокинов, по-видимому, играющих важную роль в патогенезе АП [3].
Цитокины — это регуляторные белки, которые образуют универсальную систему медиаторов, характерную как для иммунной системы, так и для клеток других органов и тканей. Под их контролем протекают все клеточные события: пролиферация, дифференцировка, апоптоз, специализированная функциональная активность клеток [6]. Термин «цитокины» был предложен N. Cohen в 1974 г., когда считалось, что они вырабатываются клетками иммунной системы, являясь одновременно и ее регуляторами. В последние годы появились данные, будто продуцентами цитокинов могут быть и эндотелиальные клетки, причем вырабатываемые ими цитокины также участвуют в регуляции процессов гемопоэза, хемотаксиса лейкоцитов, дифференцировке иммунокомпетентных клеток, синтезе острофазных белков [4].

Существует несколько вариантов классификации цитокинов в зависимости от принципа, положенного в их основу. Медиаторы можно систематизировать: а) по строению; б) по биохимическим и биологическим свойствам; в) по типам рецепторов, посредством которых они осуществляют свои биологические функции; г) в зависимости от типа клеток иммунной системы, продуцирующих данные белки (интерлейкины, моно- и лимфокины) [7].
Большинство авторов придерживаются классификации цитокинов, основанной на их биологическом действии:
Интерлейкины (IL1–IL18) – секреторные регуляторные белки, обеспечивающие медиаторные взаимодействия в иммунной системе и ее связь с другими системами организма.
Интерфероны (IFN-α, -β, -γ) – противовирусные цитокины с выраженным иммунорегуляторным действием.
Факторы некроза опухоли (TNF-α, -β) – цитокины с цитотоксическим и регуляторным действиями.
Колониестимулирующие факторы (G-CSF, M-CSF, GM-CSF) – стимуляторы роста и дифференцировки гемопоэтических клеток, регулирующие гемопоэз.
Хемокины (IL8, IL16) – хемоаттрактанты для лейкоцитов.
Факторы роста – регуляторы роста, дифференцировки и функциональной активности клеток различной тканевой принадлежности (фактор роста фибробластов, фактор роста эндотелиальных клеток, фактор роста эпидермиса) и трансформирующие факторы роста (TGF-β).
Как упоминалось выше, в основе АП лежит реакция воспаления, а цитокины являются одним из главных клеточных ответов организма на внедрение патогенной микрофлоры в ткани пародонта. Нейтрофилы и макрофаги – наиболее значимые источники медиаторов воспаления при АП, однако весьма существенную роль отводят также цитокинам и хемокинам, которые продуцируются активированными Т- и В-лимфоцитами, инфильтрированными в воспаленные ткани пародонта [8]. Деструкция соединительной и костной ткани является опосредованным результатом действия этих регуляторных белков. В литературе встречаются различные данные о роли цитокинов в развитии АП. Самые первые работы были посвящены выявлению и оценке уровней цитокинов в тканях пародонта, а в настоящее время анализируются полиморфные позиции генов различных цитокинов.
Целью данной работы являлся SNP-анализ 6 цитокинов (табл. 1) для выявления их ассоциативной связи с развитием АП.

Материал и методы
Клиническое исследование проведено на базе отделения пародонтологии ФГБУ ЦНИИС и ЧЛХ Минздрава Российской Федерации.
В исследовании принял участие 171 пациент в возрасте от 18 до 45 лет без тяжелых общесоматических заболеваний. В их число вошли 48 пациентов с диагнозом АП и 123 практически здоровых лиц, не предъявлявших никаких жалоб и без видимых клинических проявлений воспалительных заболеваний пародонта. Всем без исключения пациентам для ознакомления была предоставлена информация о проводимом исследовании в письменной и устной формах, а также форма информированного согласия для подписи на участие в исследовании.
Критерий включения: лица обоего пола, принадлежащие к европеоидной расе, в возрасте от 18 до 45 лет, проживающие на территории Москвы и Московской области.
Критерии исключения:
- системные заболевания соединительной ткани;
- злокачественные заболевания, курсы химии- и лучевой терапии в анамнезе;
- острые инфекционные и вирусные заболевания;
- поливалентная аллергия;
- беременность и лактация;
- лица, не понимающие цели исследования и не подписавшие добровольного информированного согласия.
В качестве биоматериала для проведения молекулярно-генетических исследований использована слюна, а в качестве метода – полимеразная цепная реакция (ПЦР) «в реальном времени» с использованием примыкающих флуоресцентно меченных проб (kissing probes). Лабораторная часть исследования проведена на базе ЗАО «НПФ ДНК-Технология».
Генотипирование образцов геномной ДНК
При конструировании тест-систем для картирования SNP (single nucleotide polymorphisms) использован принцип «примыкающих зондов». Принцип основан на анализе кривых зависимости интенсивности флуоресценции от температуры плавления при использовании аллель-специфичных зондов, меченных соответственно флуорофорами и гасителями. Для мечения флуорофором применено два зонда, активный концевой нуклеотид каждого из которых соответствовал одному из двух возможных аллельных вариантов геномной ДНК человека. Зонд, меченный гасителем, соответствовал инвариантной последовательности, примыкающей к полиморфной позиции.
Для установления аллельного состояния полиморфных позиций в пределах отобранных для анализа генов было использовано восемь наборов олигонуклеотидов. Из них семь наборов были использованы в практике стоматологии впервые, а один заимствован с некоторыми модификациями из докторской диссертации О.А. Зориной [2].
Результаты и обсуждение
С момента открытия хемокинов в 1980-х гг. их свойства и функциональные особенности продолжают активно изучаться. Основной функцией этого семейства белков является активация нейтрофилов и моноцитов, привлечение этих клеток в очаг воспаления. Поскольку АП – это прежде всего заболевание воспалительного характера, в наше исследование был включен ряд SNP, кодирующих некоторые белки этого семейства.
Среди отобранных для исследования генетических полиморфизмов семейства хемокинов достоверные данные были получены нами для рецептора гена лептина LEPR в позиции rs1137101 (табл. 2, 3). Частота встречаемости гетерозиготного генотипа AG гена LEPR в выборке пациентов с АП составила 41,7 %, что оказалось в 2 раза выше, чем в группе контроля. При расчете относительный риск по данному генотипу имел значение 2,42 (95 % доверительный интервал [ДИ] – 1,19–4,94), что говорит о его негативном влиянии на прогноз развития заболевания.
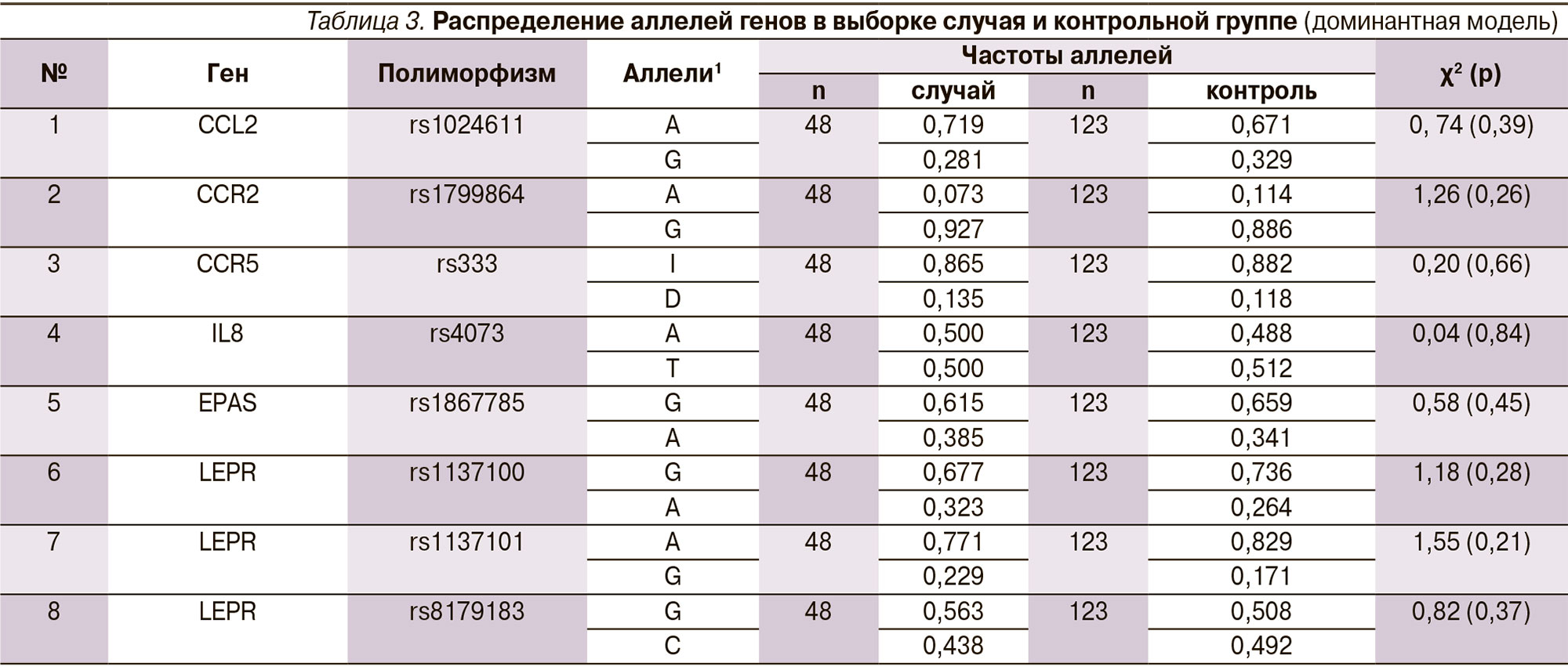
О взаимосвязи данного полиморфизма с воспалительными заболеваниями пародонта каких-либо данных в литературе нам не встречалось. Однако имеется много данных, касающихся роли этого гена в развитии других заболеваний. Часть исследований посвящена возможному риску развития ожирения. Так, в работе J.G. Gregoor и соавт. было показано, что полиморфизм LEPR Q223R (rs1137101) может быть связан с ожирением у женщин с диагнозом «психотическое расстройство», получавших атипичные антипсихотические препараты, и подчеркивается важность гендерной стратификации при исследовании влияния вариаций LEP- и LEPR-генов на метаболические побочные эффекты антипсихотических лекарств [14]. Сходное сообщение было сделано и другим коллективом авторов, показавших, что у носителей 223Q аллели LEPR были значительно более высокие масса тела (р = 0,0009) и индекс массы тела (р = 0,0022), чем при наличии 223R гомозиготы; кроме того, у 223Q-носителей значительно более высоким был риск ожирения (р = 0,0222) [13].
В работе L.Q. Wang и соавт. анализировалось влияния полиморфизма по маркеру rs1137101 в гене рецептора лептина LEPR на риск развития рака молочной железы (РМЖ) [19]. Тот же маркер использовался в нашей работе. Исследование проведено методом ПЦР «в реальном времени» на выборке из 4644 больных РМЖ и 5485 здоровых женщин. Параллельно была исследована значимость полиморфных позиций rs1137100, rs8051542, rs8051542 и rs8051542, расположенных в том же гене. Маркер rs1137101 оказался наиболее значимым: при использовании «контрастной» модели относительный риск (ОР) для выборки в целом составил 0,7 (95 % ДИ – 0,551–0,997). При этом для азиатской подгруппы в пределах выборки наиболее адекватной оказалась доминантная модель (ОР = 0,537; 95 % ДИ – 0,370–0,781). Для африканской подгруппы более достоверные различия также были обнаружены с помощью доминантной модели (OР = 1,595; 95 % ДИ – 1,207–2,108).
В целом для этой подгруппы роль полиморфизма по позиции rs1137101 гена LEPR оказалась более значимой, чем для азиатской. Контрастная модель дала для ОР = 0,716 (95 % ДИ – 0,595–0,861), а гомозиготно-кодоминантная модель – ОР = 0,537 (95 % ДИ – 0,370–0,781). Таким образом, относительно редкий аллель А в позиции rs1137101 гена LEPR оказался неблагоприятным фактором с точки зрения развития как РМЖ, так и АП.
В случае маркера rs1137100 в том же гене LEPR эффективными оказались «контрастная» модель (ОР = 0,666; 95 % ДИ – 0,603–0,720) и гомозиготно-кодоминантная модель (OР = 0,344; 95 % ДИ – 0,282–0,421, наличие аллеля G в этой позиции является неблагоприятным). Таким образом, значимость данной позиции существенно уступает rs1137101. Для остальных исследованных позиций в гене LEPR: rs8179183, rs4655537 и rs3762274, значимого влияния на вероятность развития РМЖ обнаружить не удалось.
В работе C.М. Phillips и соавт. исследовалась роль полиморфизма в гене LEPR с точки зрения риска развития инсулинорезистентности – важнейшего компонента метаболического синдрома [17]. Авторы исследования, пользуясь методами метасеквенирования, анализировали частоты встречаемости аллелей в позициях rs10493380, rs1137100, rs1137101, rs12067936, rs1805096, rs2025805, rs3790419, rs3790433, rs6673324 и rs8179183. Была собрана и исследована выборка, состоявшая из 1754 пациентов. Наиболее значимой оказалась полиморфная позиция rs3790433, наиболее распространенное аллельное состояние которой – гомозигота GG – ассоциировалось с ОР = 1,65 (95 % ДИ – 1,05–2,57; р = 0,028) по сравнению с относительно редким аллелем А, находившимся в гомо- или гетерозиготном состоянии. У пациентов, готозиготных по аллелю G, в данном положении обнаружен повышенный риск аномального подъема концентрации инсулина в плазме крови (ОР = 2,40; 95 % ДИ – 1,28–4,50; р = 0,006) и инсулинрезистентности (OР = 2,15; 95 % ДИ – 1,18–3,90; р = 0,012). Маркер rs1137101, проявивший себя в качестве значимой детерминантны риска развития агрессивного пародонтита, влияния на риск развития метаболического синдрома не оказывал.
Однако наиболее интересным сообщением по рассматриваемой тематике стало исследование восприимчивости детей к Entamoeba histolytica [18]. Дизентерийная амеба (E. histolytica) – вид паразитических простейших класса саркодовых, вызывающая тяжелое заболевание – амебиаз (амебную дизентерию, амебный колит). Было установлено, что повышенная восприимчивость к кишечной инфекции связана с заменой аминокислоты рецептора цитокина в области гомологичного домена 1 LEPR. Оказалось, что аллель G является неблагоприятным фактором развития амебиаза. Показано, что E. histolytica убивает клетки хозяина путем активации каспазы-3, что приводит к апоптозу клеток и способствует амебной инвазии [15]. Экспрессия рецептора лептина, как известно, защищает как иммунные, так и эпителиальные клетки от апоптоза вследствие воздействия раздражителя [10].
Нейтрофилы являются важнейшими клетками иммунной системы, которые представляют первую линию защиты организма от вторжения микроорганизмов. Лептин не только играет ключевую роль в регулировании массы тела, но и воздействует на другие биологические функции: гемопоэз, ангиогенез, иммунные реакции [11]. Ряд авторов приводят данные о том, что продукция лептина резко увеличивается у пациентов с сепсисом и другими воспалительными заболеваниями [9, 16]. Лептин способствует оксидативному стрессу, экспрессии адгезивных молекул, стимулирует пролиферацию и миграцию эндотелиальных и гладкомышечных клеток. Он также активирует такие воспалительные клетки, как макрофаги, нейтрофильные гранулоциты и Т-лимфоциты, стимулируя в них секрецию цитокинов [12]. Функции лептина опосредуются через рецепторы, которые экспрессируются в различных клетках и тканях, включая почки, легочную ткань, надпочечники, гемопоэтические клетки-предшественники и костный мозг, а также в нейтрофилах, моноцитах и Т-клетках.
Таким образом, можно предположить, что ген, кодирующий рецептор лептина LEPR в позиции rs1137101, может опосредованно влиять на восприимчивость индивидуума к пародонтопатогенам, провоцирующим развитие АП, по аналогии с развитием амебиаза, вызванным E. histolytica. Дальнейшие исследования этого направления, вероятно, могут пролить свет на взаимосвязь рецептора лептина с развитием АП.
В совокупности приведенные данные исследований о роли рецептора лептина в развитии заболеваний не позволяют построить модель функционирования цитокиновой системы, инициирующей деградацию соединительнотканного матрикса. Однако они подтверждают высокую информативность анализа полиморфизмов в этом гене, обусловленную высокой степенью гетерозиготности по соответствующим локусам.
