Окончание срока патентной защиты на многие оригинальные биопрепараты первой генерации стало ключевым фактором разработки и широкого выхода на фармацевтический рынок биосимиляров – препаратов, которые представляют собой воспроизведенные версии оригинальных биотехнологических лекарственных средств (ЛС).
Проблемы, связанные с использованием этого нового класса ЛС, имеют непосредственное отношение к деятельности диабетологов и эндокринологов, поскольку очень многие ежедневно используемые в нашей практике препараты – инсулины и их аналоги, гормон роста (ГР), эритропоэтины (ЭПО) – относятся к генерации биотехнол огических.
Определения
Согласно определению, указанному в первых документах Европейского фармацевтического кодекса, выделивших биосимиляры в качестве отдельного класса препаратов, биосимиляр, или “подобный биологический лекарственный продукт” (“similar biological medicinal product”), – это воспроизведенное при помощи биотехнологий ЛС, сходное с оригинальным биотехнологическим ЛС и представленное
на регистрацию после истечения срока действия патента оригинального ЛС [1]. В свою очередь биотехнологическим (или биофармацевтическим) называют препарат, который содержит активное вещество, вырабатываемое или извлекаемое из биологического источника, полученное при помощи
биотехнологических методов [1], наиболее известным из которых является метод рекомбинантной ДНК.
Характеристика биотехнологических препаратов
Действующей (активной) субстанцией биотехнологических ЛС является белок. Именно в силу особых свойств белковых молекул [2–6], которых нет у небиологических веществ, класс биотехнологических ЛС принципиально отличается от обычных химических препаратов.
Рисунок 1. Размер и структура различных белков по сравнению с молекулой химического вещества (аспирина).
Большая молекулярная масса (рис. 1), сложная и очень легкоповреждаемая пространственная структура (рис. 2), посттрансляционная модификация молекулы, нестабильность белковых молекул – все эти свойства определяют наличие высокой вариабельности как структурных, так и функциональных характеристик биотехнологических ЛС.
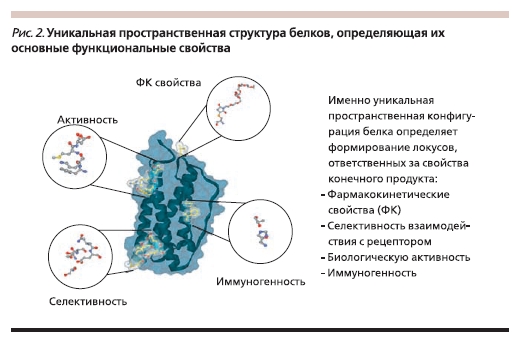
Белковые молекулы по своей природе нестабильны, их свойства могут существенно меняться вплоть до полной потери биологической активности под воздействием множества факторов. Изменение технологии на любом этапе производства – от исходных компонентов до выделения и очистки,
взаимодействие с вспомогательными субстанциями (например, стабилизатором), условия хранения – может повреждать белок и изменять его свойства.
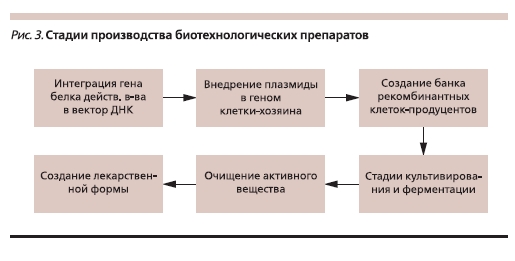
Специфика биотехнологического производства
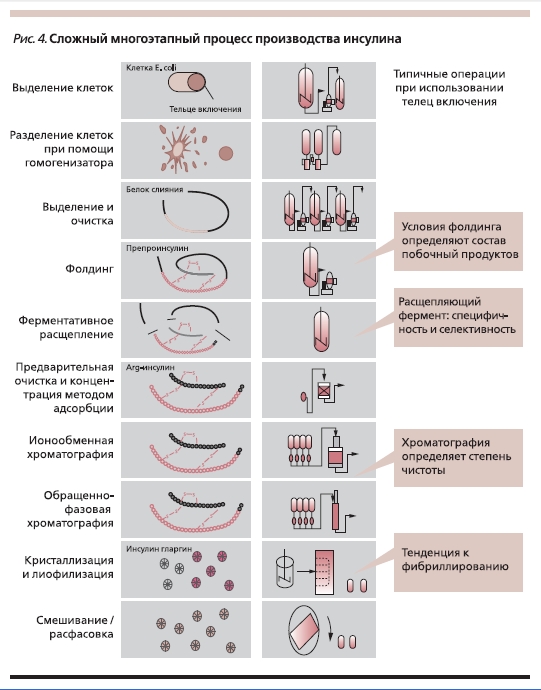
Характеристика свойств белков и производства биотехнологических препаратов (рис. 3), в т. ч. инсулинов (рис. 4), представлена в нашем предыдущем обзоре [7], поэтому мы не будем останавливаться на этом подробно. Достаточно сказать, что уже в самом термине “биосимиляры” заключены два важнейших для понимания данной концепции ключа: 1)”био” – живой, 2) “симиляр” – подобный, похожий.
Рисунок 4. Сложный многоэтапный процесс производства инсулина.
Поскольку процесс биотехнологического производства осуществляется в “живых системах” клеток (бактериях, вирусах, дрожжевых грибах), подверженных естественной и неустранимой вариабельности [3–5, 8, 9], то и продукт данного производства неизбежно вариабелен – свойство, которое получило название микрогетерогенности белков. Микрогетерогенность может проявляться даже между различными сериями препарата одного и того же производителя. Чтобы нивелировать влияние этого природного свойства белков, все используемые ингредиенты, технологические процессы, состав примесей при производстве оригинальных биопрепаратов проходят очень жесткий, многоэтапный контроль качества, обеспечивая высочайший уровень стандартизации
конечного продукта. Насколько производители биосимиляров, ориентированные на снижение стоимости и затрат на производство, могут обеспечить соответствующие стандарты качества – вопрос, который, к сожалению, не имеет однозначного ответа.
Однако даже при условии соблюдения контроля качества биосимиляр не может быть “идентичной копией” оригинального биопрепарата в силу особенностей биотехнологического производства [3–5]. Так, разработанные линии рекомбинантных клеток – продуцентов действующего вещества, как и все последующие стадии культивирования и ферментации, абсолютно уникальны для каждого биопрепарата и являются собственностью производителя оригинального бренда, т. е. предметом патента. Таким образом, существование различий между биосимилярами и оригинальными биопрепаратами неизбежно и является закономерным следствием невозможности точного воспроизведения технологии производства.
Самое важное, что последствия этих различий, особенно в отношении иммуногенности, невозможно прогнозировать [8, 10, 11]. Дело в том, что в отличие от простых молекул химических веществ, где каждый атом несет строго определенную функциональную нагрузку, структурно-функциональные взаимоотношения белков известны лишь частично [3]. При этом доступные в настоящее время лабораторные методы анализа белков не всегда обладают достаточной чувствительностью для детекции изменений их структуры и свойств. Таким образом, единственным методом достоверной оценки биотехнологических продуктов, эффективности и безопасности их применения являются
сравнительные клинические исследования [2–5, 8, 9, 12].
Неэквивалентность оригинальных и воспроизведенных
биологических препаратов
Проведенные к настоящему времени исследования убедительно доказали неэквивалентность оригинальных и воспроизведенных биологических препаратов [11, 14–19].
Это опыт компании “Мarvel”(Mumbai, India), заявка которой на регистрацию в Европе трех биосимиляров человеческого инсулина была отклонена в 2007 г. вследствие ряда недопустимых нарушений контроля качества, в т. ч. недостаточных данных о содержании примесей и иммуно-
генности препаратов. При проверке регистрационного досье были выявлены многочисленные нарушения требований к клиническим исследованиям (отсутствие контроля уровня эндогенного инсулина), а также значимые отличия фармакокинетических (ФК) и фармакодинамических
(ФД) характеристик (длительности действия, Сmax, AUC – Area under the curve) и клинической эффективности (уровня гликозилированного гемоглобина – HbA1c) по сравнению с референсным оригинальным инсулином [14–16]. При анализе биосимиляров ГР была обнаружена особая изоформа гормона, которая нарушает взаимодействие с рецептором и приводит к снижению биологической
активности. Эта изоформа образуется только при определенных условиях (высоких значениях рН и температуры), что свидетельствует о грубых нарушениях технологического процесса [17]. Крупное исследование биосимиляров ЭПО, проводившееся в трех независимых лабораториях, продемонстрировало, что состав и биологическая активность копий отличаются от оригинального препарата [18]. В частности, у 21 из 47 изученных образцов выявлено несоответствие по рН и осмолярности, у 35 – дополнительные щелочные изоформы, у 8 – превышение нормы содержания гормона, у 29 – агрегаты пептида, в 2 препаратах обнаружен эндотоксин, у 15 образцов выявлено изменение профиля активности. Другие исследования подтверждают, что биологическая активность ЭПО различных производителей может варьироваться от 70 до 200 % по сравнению с установленным стандартом [19], что в случае препарата с таким узким целевым диапазоном может иметь очень серьезные клинические последствия.
Регуляторные аспекты регистрации биосимиляров
В отличие от оригинальных ЛС (как химических, так и биотехнологических), для регистрации которых наряду с полным спектром доклинической оценки требуется предоставление данных клинических исследований, подтверждающих эффективность и безопасность терапии, для регистрации генериков
(воспроизведенных химических препаратов) достаточно доказательства биоэквивалентности, т. е. одинакового фармакокинетического профиля с оригинальным препаратом [20, 21].
В отсутствие дифференцированного подхода к воспроизведенным биологическим препаратам существует опасность применения данной “сокращенной” процедуры и для регистрации биосимиляров, что и происходит в странах, где соответствующий закон не принят. Поскольку клинические исследования являются единственным достоверным методом оценки биопрепаратов, исключение их из списка регистрационного пакета недопустимо и может повлечь за собой серьезные и непредсказуемые последствия для здоровья пациентов. Это в полной мере подтверждают данные клинических исследований. Так, в Таиланде, где по упрощенной процедуре регистрации для генериков были лицензированы и допущены к применению 14 биосимиляров ЭПО; отмечено серьезное
повышение частоты различных побочных реакций, в т. ч. снижение эффективности антианемической терапии. При оценке показателей анализов данных пациентов было установлено, что причиной резкого снижения уровня гемоглобина (в среднем до 56 г/л) является иммуногенная реакция на биосимиляр с образованием нейтрализующих антител к ЭПО и развитием красноклеточной аплазии, подтвержденной данными биопсии костного мозга [11].
Сложившаяся ситуация требует дифференцированного подхода к регистрации воспроизведенных биологических препаратов и пересмотра стандартов их допуска к клиническому применению.
Регуляторные директивы ЕМА (Европейского медицинского
агентства по лекарственным препаратам)
В Европе, где проблема биосимиляров изучается более 10 лет, уже создана мощная нормативно-правовая база, направленная на предупреждение допуска на фармакологический рынок биосимилярных копий, не доказавших эквивалентность оригинальным препаратам по эффективности и безопасности терапии.
Основные документы ЕМА, регулирующие обращение биосимиляров, – это общее руководство по подобным биологическим лекарственным продуктам 2005 г. (Guideline on Similar Biological Medicinal Products, EMEA/CHMP/437/04) [22], отдельные руководства, содержащие требования к проведению доклинических и клинических исследований биосимиляров [23], контролю анализа качества [24],
оценке иммуногенности [25], а также ряд приложений, регламентирующих регистрацию различных классов биотехнологических ЛС, в т. ч. биосимиляров инсулина [26].
Регуляторные директивы ЕМА отражают тот факт, что в силу специфики биопроизводства биосимиляр не является “идентичной копией” оригинального биотехнологического ЛС и представляет собой не аналог, а именно другой биологический препарат.
Соответственно, требования ЕМА к регистрации биосимиляров приравниваются к требованиям регистрации оригинальных ЛС и включают полный пакет документов: характеристику состава и свойств препарата, технологии производственного процесса и методов контроля качества, данные
доклинических исследований (в т. ч. токсичность на животных моделях), данные клинических исследований эффективности и безопасности терапии с обязательной оценкой иммуногенности, а также долгосрочный план управления рисками (фармаконадзор).
Требования ЕМАк биосимилярам инсулинов
В декабре 2012 г. опубликована обновленная версия руководства по оценке биосимилярных медицинских продуктов, включившего не только инсулины, но и аналоги инсулинов: “Guideline on non-clinical and clinical development of similar biological medicinal products containing recombinant human insulin and insulin analogues” [27]. Этим руководством четко определены перечень и требования к проведению доклинических и клинических исследований инсулинов, а также обоснован выбор репрезентативной популяции для их адекватной оценки.
Выбор популяции обусловлен необходимостью предупреждения влияния на результаты исследований эндогенной секреции инсулина. С этой целью в исследования обязательно должны включаться пациенты с сахарным диабетом (СД) 1 типа, которым, тем не менее, проводится контроль уровня с-пептида. К здоровым волонтерам должны применяться методы подавления собственной продукции инсулина (при помощи экзогенного инсулина или соматостатина), адекватность супрессии оцениваться
посредством мониторирования уровня с-пептида в течение всего периода исследования. Вследствие возможного влияния на чувствительность к инсулину гормонального фона у женщин рекомендовано включение в исследования только лиц мужского пола. Единственным методом достоверной оценки действия инсулинов признана стандартизованная методика гиперинсулинемического эугликемического
клэмпа, предпочтительно автоматизированная (биостатор).
Исследования, обязательные для доказательства эквивалентности эффектов исследуемого и референсного инсулина:
1. Оценка фармакокинетики (ФК) – как минимум одно однодозовое перекрестное, предпочтитель-
но двойное слепое исследование с использованием подкожного пути введения здоровым волонтерам с
нормальной массой тела и пациентам с СД 1 типа.
2. Оценка фармакодинамики (ФД) –обязательно двойное слепое перекрестное исследование с использованием гиперинсулинемического эугликемического клэмпа, позволяющее демонстрировать профиль гипогликемического ответа по соотношению “время–концентрация” и “время–эффект”. При этом необходимо предоставление информации о скорости инфузии глюкозы и концентрации инсулина. Имеет наибольшее значение для подтверждения эквивалентности эффектов исследуемого и референсного инсулина.
3. Клиническая оценка эффективности терапии – проведение отдельных исследований эффективности – признано необязательным, поскольку конечные точки подобных исследований (как правило, по уровню HbA1c) признаны недостаточно чувствительными для доказательства эквивалентности эффектов.
4. Клинические исследования безопасности терапии должны быть преимущественно сосредоточены на оценке иммуногенности продукта, тем не менее частота развития и тяжесть гипогликемий являются одними из обязательных сравниваемых конечных точек. Оцениваются наличие и титр антител при подкожном пути введения и их потенциальное влияние на гликемический контроль, потребность в инсулине и параметры безопасности, особенно реакций локальной и системной гиперчувствительности. Требуемая продолжительность исследования – не менее 12 месяцев, включая сравнительную фазу продолжительностью не менее 6 месяцев.
Успешность тактики именно законодательных мер, направленных на предупреждение допуска к применению биосимилярных копий, не доказавших эквивалентность оригинальным биопрепаратам, подтверждает продолжение истории с компанией “Marvel”, которая в 2011 г. повторила попытку зарегистрировать в ЕМА три биосимиляра инсулина (Solumarv, Isomarv, Combinarv), а в ноябре 2012
г. за месяц до выхода нового руководства самостоятельно отозвала заявку. В официальном письме “Marvel” заявила, что “решение аннулировать заявку на авторизацию препаратов связано с необходимостью подтвердить эквивалентность ФК- и ФД-свойств у пациентов с СД 1 типа при помощи
клэмп-метода, чтобы соответствовать требованиям нового руководства для инсулинов” [28]. Стоит отметить, что 11 февраля 2013 г. Комитет по лекарствам (Committee for Medicinal Products for Human Use (CHMP) Европейского Медицинского Агентства (European Medicines Agency (EMEA) опубликовал
свое повторное заключение об отказе регистрации инсулинов компании Marvel [29].
“Неинновационные копии”
инсулинов
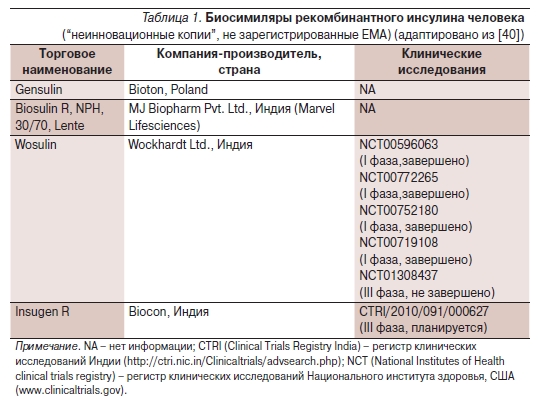
Тот факт, что до настоящего времени ЕМА не одобрено ни одного биосимиляра инсулина, указывает на несоответствие воспроизведенных инсулинов стандартам качества или как минимум недостаточную доказательную базу, чтобы это соответствие подтвердить. Тем не менее в таких странах, как Индия, Китай, Мексика, Пакистан, Перу, где не существует закона о биосимилярах, широко используются
биосимилярные копии рекомбинантного человеческого инсулина (табл. 1), а в последние 3 года появились копии аналогов инсулина (гларгина) (табл. 2).
Таблица 2. Биосимиляры рекомбинантного инсулина человека. (“неинновационные копии”, не зарегистрированные ЕМА) (адаптировано из [40]).
Помимо того что спектр исследований этих препаратов достаточно ограничен [30–33] и явно не соответствует требованиям ЕМА, установлено, что биосимиляры отличаются от оригинального гларгина по составу примесей [34–36]. Клинические последствия этих различий не изучены и, соответственно, могут представлять потенциальную угрозу, особенно в отношении иммуногенных реакций.
Следует отметить, что, согласно последним инициативам рабочей группы по биосимилярным медицинским продуктам [37], биосимилярами предложено считать только те воспроизведенные биопрепараты, которые “по данным репрезентативных сравнительных исследований продемонстрировали аналогичные свойства, эффективность и безопасность или минимальные отличия от оригинальных (референсных) препаратов, несущественные в клиническом отношении”. Все остальные воспро-изведенные биопрепараты предлагается называть “неинновационными копиями”.
На наш взгляд, подобное выделение, так сказать, “настоящих биосимиляров” не оправданно не только
потому, что значительно усложняет восприятие и без того сложной проблемы, но именно вследствие отсутствия специализированного закона о биосимилярах в большинстве стран, которые являются основными источниками “неинновационных копий”. Соответственно, абсолютно все воспроизведенные биопрепараты, зарегистрированные в таких странах, не будут соответствовать критериям ЕМА.
Фармаконадзор
В связи с ограниченностью сроков клинических исследований ряд нежелательных эффектов, в т. ч. иммуногенность, могут быть не выявлены до регистрации препарата. Поэтому для всех биосимиляров должен предоставляться план управления рисками – программа долгосрочного контроля безопасности терапии, основанная на репортировании всех нежелательных эффектов уже после регистрации препарата [23]. Однако в странах, где отсутствует закон о биосимилярах, государственная поддержка системы фармаконадзора, как правило, не работает. Кроме того, существующая система обращения ЛС по международному непатентованному наименованию (МНН) не позволяет дифференцировать биосимиляры и оригинальные биопрепараты, что представляет еще одну серьезную проблему осуществления эффективного фармаконадзора.
Заменяемость и замещение биосимиляров
Наконец вопрос, который в практическом отношении представляется ключевым: могут ли быть взаимозаменяемы оригинальные и воспроизведенные биологические препараты? Надо сказать, что понятия взаимозаменяемости и замещения ЛС близки, но не тождественны. Согласно определению Всемирной организации здравоохранения, взаимозаменяемым является препарат, который доказал терапевтическую эквивалентность с референсным препаратом сравнения [38]. Критерии терапевтической эквивалентности включают фармацевтическую эквивалентность (т. е. содержание одинакового действующего вещества в одинаковой дозе и лекарственной форме и предназначенного
для одного пути введения) и клиническую эквивалентность, т. е. равную эффективность и безопасность терапии [39]. Поскольку фармацевтически оригинальные и воспроизведенные биопрепараты отличаются a priori (вследствие особых свойств белков и различий биотехнологического производства), а клиническая эквивалентность биосимиляров не подтверждена, они не могут считаться взаимозаменяемыми.
Замещение ЛС отражает возможность замены ЛС, назначенного врачом, другим препаратом, который
считается равным по качеству, эффективности и безопасности. Эта функция отнесена к области национальной политики и регулируется на законодательном уровне [40].
В большинстве европейских стран, где действует регуляторная база ЕМА, – Франции, Германии, Греции, Италии, Словении, Испании, Швеции и Великобритании, замещение на биосимиляры запрещено. Аналогичная политика принята и в тех странах, где вступили в силу собственные законы
о биосимилярах: Канаде, Австралии, Турции, Мексике, Саудовской Аравии, Японии, Тайване, Сингапуре, Малайзии и Казахстане. В некоторых странах Европы, таких как Дания, Финляндия, Норвегия, Венгрия и Словакия, публикуется официальный перечень ЛС, которые не могут заменяться, в который включено большинство биотехнологических препаратов, в т. ч. инсулины.
В Российской Федерации до настоящего времени нет специализированных нормативно-правовых актов, регламентирующих процедуру регистрации, допуска к клиническому применению и вопросы замещения биосимиляров.
Заключение
Полученные к настоящему времени свидетельства неэквивалентности воспроизведенных и оригинальных биопрепаратов позволяют ответственно заявлять, что биосимиляры представляют особый класс ЛС, требующий дифференцированного, отличного от обычных генериков подхода. Решение о допуске биосимиляра к клиническому применению должно быть сфокусировано прежде всего на вопросах безопасности, что требует подтверждения данными собственных клинических
исследований. Поскольку доступные в настоящее время биосимиляры не соответствуют предъявляемым требованиям эквивалентности, замещение на них оригинальных препаратов преждевременно и не оправданно.

{kind=link}
{kind=link}
{kind=link}