
Обоснование
Первичная надпочечниковая недостаточность (ПНН) – тяжелое состояние, связанное с дефицитом в организме гормонов коры надпочечников (гипокортицизм). Неспецифичность клинических симптомов и относительная редкость данной патологии у детей – главные причины поздней или неправильной диагностики. Следствием недостаточности выработки кортизола являются слабость, вялость, заторможенность, пигментация кожных покровов («бронзовая кожа»), снижение аппетита и потеря массы тела, возможны судороги, снижение артериального давления (АД) и потеря сознания. Дефицит другого гормона надпочечников альдостерона приводит в первую очередь к электролитным нарушениям (гиперкалиемии и гипонатриемии) и проявляется неукротимой рвотой, диареей, обезвоживанием, болями в животе, снижением АД, нарушениями сердечного ритма [1, 2].
Аналогичные проявления могут наблюдаться при различных инфекционных заболеваниях (в т.ч. при новой коронавирусной инфекции), аппендиците, эпилепсии и других более распространенных, чем надпочечниковая недостаточность (НН), состояниях. Единственным специфичным симптомом является гиперпигментация кожных покровов, но и этот симптом может быть слабовыраженным [3].
Новая коронавирусная инфекция (COronaVIrus Disease, 2019), впервые зарегистрированная в декабре 2019 г. в Китае в провинции Ухань, стала распространяться не только среди взрослого населения, но и в детской популяции. К наиболее частым симптомам заболевания относятся стойкая лихорадка, общая и мышечная слабость, миалгии, сухой кашель с небольшим количеством мокроты. Реже наблюдаются симптомы со стороны желудочно-кишечного тракта (диспепсия, рвота, диарея) [4, 5]. Клиническое течение COVID-19 у детей может носить атипичный характер и маскировать неспецифичные симптомы других заболеваний, приводя к несвоевременной диагностике.
При ПНН наряду с физической адинамией развивается психическая астенизация вплоть до развития психозов. Мышечная слабость служит результатом нарушения углеводного (гипогликемия) и электролитного (гипонатриемия) обменов. Патогенез желудочно-кишечных расстройств объясняется снижением секреции соляной кислоты и пепсина, а также повышенной секрецией хлорида натрия в просвет кишечника. Рвота и диарея усиливают потерю натрия, что может приводить к развитию острой НН. Больные ощущают постоянную потребность в соленой и острой пище [6, 7].
ПНН ранее нередко имела приобретенный характер, развивалась в результате туберкулезного поражения надпочечников [6, 7]. На сегодняшний день в подавляющем большинстве случаев ПНН у детей вызвано наследственными синдромами. Известно более 20 наследственных заболеваний, которые приводят к развитию ПНН. К наиболее частым из них, приводящим к манифестации гипокортицизма после 3 лет у детей, относятся аутоиммунный полигландулярный синдром 1-го типа (АПС-1), Х-сцепленная адренолейкодистрофия – АЛД (заболевание проявляется только у мальчиков), синдром Оллгрова, синдром Кернса–Сейра. Определение конкретной нозологической формы ПНН позволяет прогнозировать течение заболевания, вероятность появления патологии других органов и систем и определять тактику лечения пациента [2, 3].
Клинический случай
На консультацию к детскому эндокринологу обратились родители мальчика А. 11 лет с жалобами на повышенную вялость, слабость, плохой аппетит вплоть до отказа от еды, снижение массы тела, избирательную тягу к соленой пище, лабильность настроения от плаксивости до агрессии.
Из анамнеза заболевания, со слов родителей, стало известно, что вышеперечисленные жалобы появились в марте 2021 г. на фоне перенесенной новой коронавирусной инфекции COVID-19 (подтверждена методом полимеразной цепной реакции), клиническая симптоматика расценивалась как тяжелое проявление инфекции с нейротоксикозом. Позже, в начале апреля, присоединились жалобы на многократную рвоту, боли в животе, в связи с чем ребенок был госпитализирован в инфекционное отделение с диагнозом «острый гастроэнтерит c тяжелым эксикозом 1–2-й ст.». По результатам обследования отмечалась гиперкалиемия (К 7,17 ммоль/л), гипонатриемия (Na 107,7 ммоль/л). Больной получал инфузионную, симптоматическую терапию с удовлетворительным эффектом.
Улучшение состояния носило временный характер: вскоре возобновилась рвота, и в тяжелом состоянии ребенок был госпитализирован в отделение детской реанимации и интенсивной терапии. В анализах: гликемия – 3,4–4,6–9,4 ммоль/л, калий – 3,88–4,0–3,9, Na – 138–139–139 ммоль/л. В ходе обследования пациента проведено магнитно-резонансное (МРТ)-исследование головного мозга (16.04.2021), не выявившее патологических изменений. По данным фиброгастродуоденоскопии были диагностированы недостаточность кардии, хронический гастродуоденит, дуодено-гастральный рефлюкс. Получал антибактериальную терапию (цефепим, амикацин), инфузионную терапию.
В связи с положительной динамикой состояния ребенка и выявленной гастропатологией 17.04.2021 больной переведен в гастроэнтерологическое отделение Республиканской детской больницы по месту жительства. По результатам обследования повторно зафиксирована гипонатриемия (123–119 ммоль/л), калий в пределах нормы – 4,1–5,35 ммоль/л. В гормональном профиле (20.04.2021): кортизол – 540 нмоль/л (норма –101,2–535,7), тестостерон – 0,1 нмоль/л (норма <0,8), 17-ОПН – 0,55 нг/мл (норма – 0,21–4,06), паратгормон – 5,1 пг/мл (норма – 1,6–6,9), АКТГ – 1250 пг/мл (норма <46), ренин >500 мкМЕ/мл. По данным ультразвукового исследования органов брюшной полости выявлены эхопризнаки реактивных изменений печени, деформации желчного пузыря с застойными явлениями, по данным МРТ – признаки гепатомегалии, надпочечники без патологических изменений. В отделении отмечалась невыраженная положительная динамика на фоне соблюдения диеты и лечения гастропатологии.
Из-за ухудшения состояния в 03.05.2021–19.05.2021 повторно госпитализирован в отделение гастроэнтерологии с диагнозом «постковидный синдром, реактивный гепатит, катаральный рефлюкс-эзофагит, недостаточность кардии, хронический гастродуоденит, обострение».
В связи с отсутствием эффекта от проведенной терапии и изменениями в анализах педиатр вновь инициировал консультацию детского эндокри-нолога.
Из анамнеза жизни: ребенок от 1-й беременности, протекавшей без осложнений, 1-х самостоятельных родов. Масса тела при рождении – 2800 г, длина тела – 48 см. Находился на грудном вскармливании до 6 месяцев. Развитие на первом году жизни протекало без особенностей. Аллергические заболевания родители отрицают.
 В возрасте 5 месяцев перенес закрытую черепно-мозговую травму без осложнений. Занимался профессионально спортом (грэпплинг) с 7 лет до появления вышеперечисленных жалоб.
В возрасте 5 месяцев перенес закрытую черепно-мозговую травму без осложнений. Занимался профессионально спортом (грэпплинг) с 7 лет до появления вышеперечисленных жалоб.
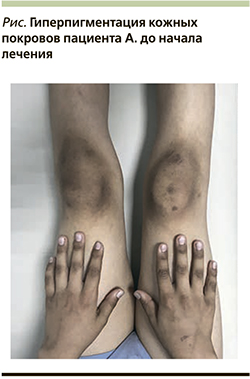
Наследственный анамнез: брат (8 лет), сестра (3 месяца), родители здоровы. Объективно: рост – 138,8 см (SDS роста -0,85), масса тела – 29,2 кг (SDS массы тела -1,35). Индекс массы тела – 15,2 кг/м2 (SDS ИМТ -1,19). Состояние средней степени тяжести. В сознании. На вопросы отвечает односложно. Отмечается лабильность настроения от плаксивости до повышенной эмоциональности. Телосложение астеническое. Подкожная клетчатка развита недостаточно, распределена равномерно. Кожные покровы смуглые, чистые, суховатые. Отмечена очаговая гиперпигментация над мелкими суставами кистей рук, над локтевыми и коленными суставами (см. рисунок). Слизистые оболочки бледно-розовой окраски, чистые. Периферические лимфатические узлы не увеличены, не спаяны между собой, при пальпации безболезненны.
Форма грудной клетки не изменена. Над всей поверхностью легких выслушивается везикулярное дыхание, хрипов нет. Область сердца визуально не изменена. Тоны сердца ясные, ритм правильный. Частота сердечных сокращений (ЧСС) – 78 в минуту. АД – 110/70 мм рт.ст. Живот мягкий, безболезненный. Печень выступает из-под края реберной дуги на +1 см. Стул регулярный, оформленный. Диурез адекватный. Щитовидная железа не увеличена, при пальпации умеренно эластичной консистенции, клинически эутиреоз. В позе Ромберга устойчив. Половое развитие по мужскому типу: Таннер 1 (G 1, P 1), тестикулы в мошонке, d=s=5 мл. Со стороны костно-мышечной системы патологических изменений не выявлено.
На основании осмотра высказано предположение, согласно которому у ребенка имеет место ПНН и рекомендовано повторное исследование электролитов крови и гормонального профиля для уточнения генеза.
В течение 2–3 дней после данной консультации (25.05.2021) в связи с ухудшением состояния, возобновлением тошноты, выраженных болей в животе и усиливающейся слабости ребенок вновь госпитализируется в отделение реанимации и интенсивной терапии с клиникой токсикоза 2-й степени на фоне водно-электролитных нарушений. После стабилизации состояния больной переведен в эндокринологическое отделение. В биохимическом анализе крови: гипонатриемия (122,1–105,9–117,3 ммоль/л), калий в пределах 3,67–4,78–3,54 ммоль/л (норма – 3,4–4,7), кальций – 2,05–2,6 ммоль/л (норма – 2,20–2,70). В гормональном профиле от 29.05.2021 выявлены признаки гипокортицизма: кортизол – 27,3 нмоль/л (норма – 101,2–535,7), тестостерон – 0,1 нмоль/л (норма <0,98), 17-ОПН – 0,18 нг/мл (норма – 0,21–4,06), адренокортикотропный гормон (АКТГ) более 1250 пг/мл (норма <46 пг/мл).
Проведена мультиспиральная компьютерная томография забрюшинного пространства с контрастным усилением: патологических образований и изменений надпочечников выявлено не было.
На основании клинической картины и полученных результатов дополнительных исследований (гиперкалиемия, гипонатриемия, высокие уровни АКТГ и ренина, пониженный уровень кортизола) установлен диагноз «первичная надпочечниковая недостаточность». Назначена заместительная гормональная терапия: гидрокортизон (Кортеф) –15 мг/сут, флудрокортизон (Кортинефф) – 0,1 мкг/сут.
На фоне проводимой терапии в стационаре у ребенка возникли зрительные галлюцинации, резкие перепады настроения, которые пришлось верифицировать со следующими состояниями: с побочным действием флудрокортизона в виде психических реакций, которые могут имитировать шизофрению, мании или делириозного синдрома, с «Аддисоновой» энцефалопатией, характеризующейся психозами и лицевыми гримасами на фоне некомпенсированной НН и выраженным эксикозом, с неврологической симптоматикой при АЛД. Флудрокортизон был отменен в связи с нормализацией электролитов крови и появлением отечности, а также с целью дифференциальной диагностики галлюцинаций. Консультация психиатра в стационаре профильных заболеваний у ребенка не выявила.
После стабилизации состояния ребенок направлен в ФГБУ НМИЦ (Национальный медицинский исследовательский центр) эндокринологии Минздрва РФ для уточнения генеза ПНН.
Результаты дополнительных исследований в ФГБУ НМИЦ МЗ РФ, Москва (18.06.2021): в общем анализе крови выявлен тромбоцитоз (тромбоциты – 426×109 кл/л). Биохимические и гормональные показатели представлены в табл. 1, 2. Обращают на себя внимание высокий уровень ренина и АКТГ, гипонатриемия.

По данным электрокардиографии, ритм синусовый с ЧСС – 72–74 уд/мин.
Электрическая ось сердца отклонена влево. Изменение предсердного компонента. Неполная блокада левой передней ветви и правой ножки пучка Гиса.
Имелось предположение о наличии у ребенка ахалазии кардии из-за частой рвоты и дисфагии в анамнезе, однако результаты рентгеноскопии пищевода не подтвердили ее наличия и тем самым исключили наличие синдрома Оллгрова как одной из причин НН в рамках наследственных синдромов.
В связи с отсутствием данных МРТ и компьютерной томографии органов брюшной полости и забрюшинного пространства в пользу туберкулезного или другого характерного поражения надпочечников с целью уточнения генеза заболевания и исключения Х-сцепленной АЛД инициировано молекулярно-генетическое исследование и исследование уровня очень длинноцепочечных жирных кислот (ОДЦЖК). В ожидании результатов дополнительных исследований ребенок выписан под наблюдение детского эндокринолога по месту жительства на заместительной гормональной терапии: гидрокортизон в дозе 15 мг/сут и флудрокортизон 0,1 мг/сут.
Обсуждение
НН может встречаться как изолированная форма на фоне туберкулезного или аутоиммунного поражения надпочечников, так и в рамках других заболеваний (Х-сцепленная АЛД, синдром Оллгрова, аутоиммунный полигландуллярный синдром 1-го типа), которые в процентном соотношении доминируют в детском возрасте [2, 3, 6].
Х-сцепленная АЛД (болезнь Зимерлинга–Крейтцфельда, меланодермическая лейкодистрофия) – наследственное заболевание с X-сцепленным рецессивным типом наследования, относящееся к группе пероксисомных болезней и проявляющееся преимущественно поражением белого вещества нервной системы и коры надпочечников. Болезнь обусловлена дефектами гена ALD (22q28), проявляющимися недостаточностью лигноцероил-КоФ-лигазы. Это в свою очередь ведет к нарушению р-окисления насыщенных длинноцепочечных жирных кислот (ДЦЖК), имеющих 24–32 атома углерода, в пероксисомах и последующему их накоплению вместе с эфирами холестерина в клетках нервной системы и коркового вещества надпочечников в виде слоистых, триламинарных внутриклеточных включений, нарушению β-окисления жирных кислот в пероксисомах, которое ведет к накоплению ДЦЖК. Частота этого заболевания составляет 1 случай на 20 тыс. рождений. Клинически выделяют несколько вариантов АЛД. Церебральная форма («классическая» АЛД), манифестирующая в детском возрасте (5–12 лет), самая распространенная и тяжело протекающая разновидность этой патологии (45% случаев). В 86% случаев неврологические и психические расстройства часто предшествуют клиническим и лабораторным признакам НН. Наиболее часто основными симптомами в этом возрасте являются гиперактивное или, наоборот, аутистическое поведение, эпизоды агрессивности, проблемы обучения, снижение памяти, дефицит внимания, прогрессирующая деменция и нарушение походки. Менее частые симптомы – это нарушения зрения и слуха, признаки НН. По мере прогрессирования заболевания развиваются спастический тетрапарез, слепота, глухота, судороги, не отвечающие на антиэпилептическую терапию [8, 9].
К методам биохимического подтверждения диагноза Х-АЛД относится выявление в плазме крови, эритроцитах, лейкоцитах, культуре клеток кожных фибробластов повышенного уровня ОДЦЖК, особенно тетракозановой (С24:0) и гексакозановой (С26:0) кислот и их соотношений С24:0/С22:0 и С26:0/С22:0 [8, 9]. Вне криза НН уровень электролитов крови и кортизол могут оставаться в пределах нормы, а AКТГ изолированно повышаться.
Результаты дополнительных исследований выявили, что у нашего пациента А. в плазме крови повышено соотношение концентраций ОДЦЖК (табл. 3). Данные изменения могли быть обусловлены особенностями питания пациента и требовали дальнейшего наблюдения.

На начальных стадиях заболевания при Х-АЛД на МРТ головного мозга имеются специфические изменения: выявляется гиперинтенсивный сигнал (в Т2W) в области мозолистого тела, кортикоспинальных и кортикопонтинных трактов, который по мере прогрессирования быстро распространяется в затылочные и задне-теменные отделы [8]. МРТ-снимки больного А., сделанные ранее по месту жительства, были пересмотрены специалистами ФГБУ НМИЦ Минздрава РФ и патологии, характерной для АЛД, выявлено не было.
Результаты молекулярно-генетического исследования показали, что в гене SLC12A3 (NM 000339.3) в 6-м экзоне обнаружена замена одного нуклеотида в гетерозиготном состоянии с.775G>A, приводящая к замене аминокислоты p.D259N с глубиной покрытия 106х (rs780461639). Вариант очень редко встречается в базах данных аллельных вариантов человека и описан в литературе как патогенный при синдроме Гительмана. По совокупности данных найденная замена расценивается как патогенная. Другие редкие варианты в гене SLC12A3 не найдены, кодирующая часть и прилежащие участки интронов покрыты полностью. При аутосомно-доминантном типе наследования обнаружения одного варианта в гене недостаточно для объяснения причины заболевания на молекулярно-генетическом уровне.
Синдром Гительмана – это наследственная тубулопатия, характеризующаяся синдромом метаболического алкалоза. Заболевание имеет аутосомно-рецессивный тип наследования, манифестирует у детей школьного возраста выраженной гипомагниемией, гипокалиемией, метаболическим алкалозом, судорогами конечностей, гипокальциурией, отсутствием нефрокальциноза, полиурией и никтурией, нормальной концентрационной функцией почек [10]. Распространенность синдрома Гительмана – 1:40 000–50 000 [11].
С целью исключения данного заболевания ребенку вновь были проведены дополнительные исследования, не выявившие патологических отклонений. Биохимический анализ крови от 25.10.2021: магний – 0,9 ммоль/л (норма – 0,7–0,86), натрий – 145,31 (норма – 136–145), калий – 3,93 ммоль/л (норма – 3,5–5,1), ренин – 5,57 мкМЕ/мл (норма – 4,4–46,1). Кальций в суточной моче – 2,75 ммоль/сут (норма – 0,0–8,8).
В ходе дифференциальной диагностики генеза НН у представленного нами пациента исключался синдром Оллгрова, или триплет A (AAAS – Alacrimia, Achalasia, Adrenal insufficiency) – редкое полисистемное заболевание, характеризующееся хронической НН, алакримией и ахалазией кардии, связанной с мутациями в гене AAAS [12, 13]. Поводом для исключения данного заболевания было сочетание НН и многократной рвоты в анамнезе у ребенка. НН – один из признаков триплета А, встречающийся в 100% случаев заболевания, однако дебютирует обычно позже других проявлений: в первом десятилетии жизни, в подростковом периоде или даже у взрослых [14]. Она развивается вследствие резистентности надпочечников к АКТГ, что приводит к значимому повышению его уровня (АКТГ) и низкому уровню кортизола в сыворотке крови.
Ахалазия кардии обычно возникает в детском возрасте, чаще в первое десятилетие жизни, но может развиваться и позже. Встречается приблизительно в 93% случаев и проявляется рвотой, дисфагией, замедленным набором массы тела и хроническим кашлем [15]. Рентгеноскопия пищевода исключила наличие данной патологии у представленного нами больного.
Алакримия – наиболее постоянный и часто встречающийся симптом (практически во всех случаях с рождения) при синдроме Оллгрова и рассматривается в качестве раннего диагностического признака заболевания [16, 17].
Синдром Оллгрова нередко ассоциируется с нейродегенерацией и неврологической дисфункцией, проявляется вегетативными нарушениями, умственной отсталостью, амиотрофией [16, 17].
Заключение
Представленный клинический случай вызывает интерес редкой встречаемостью ПНН в детском возрасте. Отсутствие характерных электролитных и гормональных нарушений в полном объеме и манифестация заболевания на фоне новой коронавирусной инфекции COVID-19 привели к ошибочной длительной дифференциальной диагностике и отсутствию адекватного лечения. Неспецифичность клинических симптомов (повышенная утомляемость, слабость, рвота, потеря массы тела) обусловила многократные госпитализации ребенка в непрофильные отделения. Тяжелое течение COVID-19 в детском возрасте не типично и должно насторожить в плане диагностического поиска.
В данном случае вирусная инфекция послужила пусковым механизмом в развертывании клинической картины ПНН и усугубила течение заболевания [18].
Исходом длительного обследования ребенка является исключение таких заболеваний, как Х-АДЛ, АПС-1, синдром Оллгрова, синдром Гительмана, туберкулезное поражение надпочечников. Планируется молекулярно-генетическое исследование биологического материала родителей с целью дальнейшей верификации генеза НН у ребенка.
В настоящее время ребенок получает заместительную терапию: гидрокортизон (Кортеф) в дозе 15 мг/сут и флудрокортизон (Кортинефф) 0,1 мг/сут под наблюдением детского эндокринолога по месту жительства с положительным эффектом.
Согласие пациента. Пациентами добровольно подписано информированное согласие на публикацию персональной медицинской информации.
Финансирование. Отсутствует.


