Введение
Адреногенитальный синдром (АГС), или врожденная гиперплазия коры надпочечников (ВГКН), является одним из самых распространенных наследственных моногенных заболеваний, одновременно представляет собой вариант хронической первичной надпочечниковой недостаточности и группу патологии полового развития, а также половой дифференцировки. Кроме того, проблема АГС в стертой (неклассической) форме занимает существенное место среди причин нарушения репродуктивного здоровья (бесплодие, невынашивание беременности). Таким образом, с проблемой АГС встречаются врачи разных специальностей: неонатологи, педиатры, эндокринологи, гинекологи, генетики. Понимание основных принципов диагностики и лечения этого заболевания врачами разных специальностей является необходимым во избежание серьезных ошибок на разных этапах оказания медицинской помощи.
Клинические варианты АГС
АГС – группа заболеваний с аутосомно-рецессивным типом наследования, в основе которых лежит дефект одного из ферментов, участвующих в синтезе кортизола.
В зависимости от фермента, в гене которого имеется дефект, на сегодняшний день известно семь нозологических вариантов АГС:
• липоидная гиперплазия надпочечников (дефект StAR-протеина);
• дефицит Р450scc (20,22-десмолазы);
• дефицит 3βГСД (3β-гидроксистероиддегидрогеназы);
• дефицит CYP17 (17α-гидроксилазы/17,20-лиазы);
• дефицит CYP21 (21-гидроксилазы);
• дефицит CYP11В1 (11β-гидроксилазы);
• дефицит POR (Р450 оксидоредуктазы).
До 95 % всех случаев АГС составляет дефицит 21-гидроксилазы. Другие нозологические формы АГС встречаются редко.
Дефект 21-гидроксилазы: классификация и клинические проявления
Ген CYP21, кодирующий фермент 21-гидроксилазу, локализован на коротком плече 6-й хромосомы. Описано более пятидесяти мутаций этого гена, приводящих к синтезу фермента со степенью активности от 0 до 60 % [1].
Частота встречаемости классических вариантов дефицита 21-гидроксилазы, рассчитываемая по данным неонатального скрининга, в разных популяциях колеблется от 1 : 10 тыс. до 1 : 18 тыс. новорожденных. Чрезвычайно высокая частота выявлена в двух изолированных популяциях: у эскимосов Западной Аляски – 1 : 280, и у жителей острова Ла Руньон в Индийском океане – 1 : 2100. Частота неклассического варианта дефицита 21-гидроксилазы значительно выше – от 0,3 до 0,01 % в мировой популяции – и достигает 3,7 % среди евреев Ашкенази. По результатам неонатального скрининга, введенного в России в 2006 г., частота данной патологии в Москве составила 1 : 10 тыс. живых новорожденных.
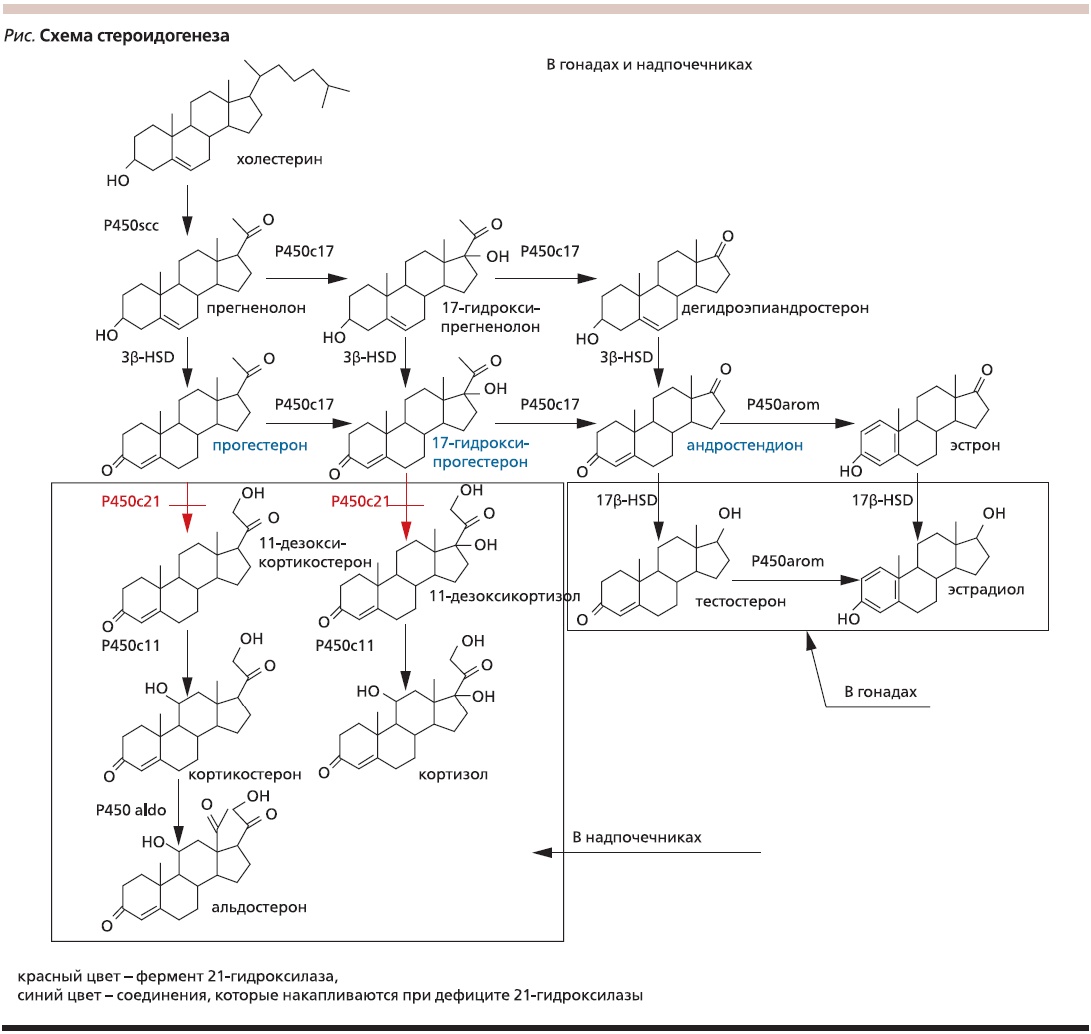
Дефект 21-гидроксилазы приводит к недостаточной продукции кортизола и ответному повышению уровня секреции адренокортикотропного гормона (АКТГ), что в свою очередь обусловливает гиперплазию коры надпочечников. Гиперплазированные надпотировать стероиды, предшествующие ферментативному блоку – прогестерон и 17-гидроксипрогестерон (17ОНП), а также андрогены, биосинтез которых не зависит от 21-гидроксилазы (см. рисунок). В результате имеется дефицит глюкокортикоидов (кортизола) и минералокортикоидов (альдостерона), а также избыток андрогенов (дегидроэпиандростерона, андростендиона, тестостерона).
Рисунок. Схема стероидогенеза.
Дефицит глюкокортикоидов (кортизола) приводит к развитию гипогликемического синдрома, что особенно тяжело проявляется у новорожденных, но также может развиваться в любом возрасте при неадекватной заместительной терапии. Развитию гипогликемии способствуют перерывы в кормлениях и интеркуррентные заболевания. Высокий уровень АКТГ, который поднимается по механизму обратной связи в ответ на низкий уровень кортизола, проявляется наличием гиперпигментаций. АКТГ – один из производных проопиемеланокортина (ПОМК)
ПОМК имеет высокое сродство к меланокортиновым рецепторам, в частности к MCR1 (рецептору меланоцитстимулирующего гормона). Большое количество АКТГ, конкурируя с меланоцитстимулирующим гормоном, связывается с его рецептором и вызывает повышение синтеза меланина в коже, что приводит к гиперпигментациям.
Дефицит минералокортикоидов (альдостерона) проявляется синдромом потери соли, который включает срыгивания, массивные рвоты “фонтаном”, полиурию, жажду, обезвоживание и низкое артериальное давление.
Избыток надпочечниковых андрогенов в эмбриональный период приводит к вирилизации наружных гениталий у плодов с кариотипом 46ХХ. Степень вирилизации наружных гениталий у девочек варьируется от 2-й до 5-й степени по шкале Прадера. После рождения избыток надпочечниковых андрогенов приводит к преждевременному половому развитию по изосексуальному типу у мальчиков и по гетеросексуальному типу у девочек. У мальчиков увеличиваются размеры полового члена, появляются эрекции. У девочки увеличиваются размеры клитора, отмечается его напряжение. К 1,5–2,0 годам у детей обоего пола появляются лобковое оволосение, угревая сыпь, грубеет голос. В первые годы жизни линейный рост ускорен, однако степень костной дифференцировки опережает рост и зоны роста закрываются к 9–10 годам, что в конечном итоге приводит к низкорослости.
Классификация дефицита 21-гидроксилазы основана на клинической картине заболевания и включает три клинические формы в зависимости от выраженности симптомов: сольтеряющую, вирильную и неклассическую. Тяжесть клинических проявлений определяется степенью снижения активности фермента 21-гидроксилазы. Минералокортикоидная недостаточность (т. е. сольтеряющая форма) развивается только при нулевой активности фермента (отмечается в 60 % всех случаев). Поскольку в норме секреция альдостерона в 1000 раз меньше секреции кортизола, уже 1 % активности фермента оказывается достаточным для поддержания водно-солевого баланса.
Самая тяжелая из них – сольтеряющая форма – проявляется с первого месяца жизни как у мальчиков, так и у девочек плохим набором веса, рвотами “фонтаном”, срыгиваниями, отсутствием аппетита; нарастают гиперкалиемия и гипонатриемия. Потеря соли приводит к выраженной дегидратации, которая усугубляется частыми и массивными рвотами. В отсутствие терапии может наступить смерть ребенка в результате коллапса и кардиогенного шока.
При сольтеряющей и вирильной формах повышенное количество надпочечниковых андрогенов внутриутробно приводит к активной вирилизации наружных гениталий. К моменту рождения наружные гениталии девочки имеют бисексуальное строение: клитор гипертрофирован, отмечается сращение мошоночного шва различной степени. В некоторых случаях внутриутробная андрогенизация выражена настолько сильно, что наружные гениталии практически полностью соответствуют мужским и девочка ошибочно регистрируется и воспитывается как мальчик. У мальчиков при рождении наружные гениталии соответствуют полу, может отмечаться небольшое увеличение полового члена.
После рождения симптомы андрогенизации у детей обоего пола нарастают. У девочек увеличиваются размеры клитора, отмечается его напряжение. У мальчиков увеличиваются размеры полового члена, появляются эрекции. Кроме того, избыток андрогенов приводит к ускорению темпов физического развития и прогрессированию темпов костного созревания.
Неклассическая форма заболевания проявляется в возрасте 4–5 лет клинической картиной преждевременного адренархе (преждевременного появления полового оволосения). Клинических проявлений надпочечниковой недостаточности при этой форме не бывает.
Диагностика дефицита 21-гидроксилазы
Главным гормональным маркером диагностики дефицита 21-гидроксилазы является повышенный уровень 17ОНП в крови [3].
Все стероидные соединения имеют схожую химическую структуру, что обусловливает значительный перекрест между ними при определении их концентрации в крови. Это может вызывать определенные трудности в диагностике формы АГС. В настоящее время максимальной информативностью обладает метод мультистероидного анализа с предшествующим разделением всех стероидов с помощью хроматографии или масс-спектрометрии. При использовании данного метода можно не только определять точную концентрацию стероидных соединений, но и оценивать соотношение предшественников и продуктов различных ферментов.
Регуляция синтеза альдостерона осуществляется ренин-ангиотензиновой системой, поэтому при сольтеряющей форме определяется повышенный уровень активности ренина плазмы.
До наступления эры неонатального скрининга новорожденных исследование гормональных маркеров АГС проводилось при наличии соответствующих симптомов. Диагностика дефицита 21-гилроксилазы проводилась новорожденным девочкам (с кариотипом 46ХХ) с неправильным строением наружных гениталий и новорожденным мальчикам с симптомами срыгивания, потерей веса, обезвоживанием, гиперкалиемией, гипонатриемией, а также мальчикам 3–4 лет с симптомами преждевременного адренархе, ускорения роста и костного возраста, у которых следует подозревать вирильную форму заболевания.
Неонатальный скрининг на АГС в России
В 2006 г. в рамках программы “Национальные приоритетные проекты” в России был введен неонатальный скрининг на АГС, в соответствии с которым исследование 17-ОНР проводится всем новорожденным на 5-е сутки жизни (недоношенным детям – позже).
Молекулярно-генетическая диагностика
Исследование гена CYP21 в настоящее время является доступным в России и частоприменяемым методом подтверждающей диагностики. Проведение молекулярно-генетического анализа и выявление мутаций в гене CYP21 позволяют определять форму заболевания, прогнозировать его течение, а также определять вероятность рождения больного ребенка, возможности пренатальной диагностики и лечения.
Генетическое консультирование
Как и при любом наследственном заболевании, родителям больного ребенка и совершеннолетнему пациенту объясняют суть имеющихся генетических нарушений и вероятность рождения больных детей в будущем.
Для всех форм АГС возможно проведение пренатальной диагностики. Пренатальную диагностику проводят в семьях, где оба родителя являются гетерозиготными носителями дефекта гена CYP21, как правило, уже имеющие детей с данным заболеванием. Принцип пренатальной диагностикисостоит в исследовании ДНК, полученной из ворсинок хориона, взятых методом пункционной биопсии на 9–11-й неделе беременности. Кроме того, в последние годы широко внедрен метод дородовой и предимплантационной диагностики АГС, который позволяет существенно повышать вероятность рождения здорового ребенка с помощью методов экстракорпорального оплодотворения.
Лечение АГС
Все пациенты с АГС получают глюкокортикоидную терапию. Препараты глюкокортикоидов возмещают дефицит собственного кортизола и по механизму отрицательной обратной связи подавляют чрезмерную секрецию АКТГ, тем самым снижая высокий уровень надпочечниковых андрогенов. Пациенты с сольтеряющей формой 21-гидроксилазной недостаточности получают дополнительно к глюкокортикоидным препаратам лекарственное средство с минералокортикоидной активностью – флудрокортизон.
Согласно международному консенсусу по диагностике и лечению 21-гидроксилазной недостаточности у детей, принятому в 2002 г. на объединенном конгрессе Европейского общества педиатров-эндокринологов и Общества педиатров-эндокринологов Лоусона Вилкинса (США), препаратом выбора для детей с открытыми зонами роста является таблетированный гидрокортизон (Кортеф), принимаемый троекратно в сутки в равных дозах и через равные промежутки времени [3]. У детей пубертатного и постпубертатного возраста возможно применение пролонгированных препаратов – преднизолона и дексаметазона. Средние дозы гидрокортизона составляют 10–20 мг/м2. Указанные дозы выше, чем физиологическая секреция кортизола: 6–7 мг/м2 – у детей старшего возраста и 7–9 мг/м2 – у новорожденных и грудных детей [4]. Применяемые супрафизиологические дозы глюкокортикоидов при лечении АГС необходимы для подавления гиперсекреции андрогенов и предупреждения развития острой надпочечниковой недостаточности. Имея короткий период полужизни, гидрокортизон оказывает минимальное супрессивное воздействие на процессы роста ребенка по сравнению с другими глюкокортикоидными препаратами.
Следует отметить, что особую сложность представляет подбор адекватной дозы глюкокортикоидов. Контроль адекватности терапии дефицита 21-гидроксилазы основан на показателях физического развития и данных гормонального обследования. Одним из основных параметров эффективности и адекватности глюкокортикоидной терапии является рост пациентов. Недостаточная доза глюкокортикоидов не подавляет АКТГ и обусловливает высокий уровень андрогенов, что проявляется высокими темпами роста, быстрым прогрессированием темпов скелетного созревания и прогрессированием признаков вирильного синдрома. Костное созревание опережает ускорение темпов роста, что приводит к низкорослости. Передозировка глюкокортикоидов также негативно влияет на рост ребенка: снижение скорости роста указывает на длительную передозировку. Об избыточной дозе могут свидетельствовать быстрая прибавка в весе, повышение аппетита. Адекватная терапия глюкортикоидными препаратами обеспечивает нормальные темпы роста и костного созревания [5]. Во всех публикациях, посвященных анализу роста пациентов с АГС, конечный рост пациентов оказывался ниже популяционного или целевого роста [6–10].
Помимо клинических данных при оценке адекватности терапии используют гормональные показатели. Основным критерием является уровень 17ОНП сыворотки крови; можно ориентироваться также на уровень андростендиона в крови и экскрецию прегнантриола с мочой. При гормональном контроле в процессе терапии следует опасаться снижения уровня 17ОНП до минимальных нормативных показателей – желательно поддерживать его в верхних пределах нормы или несколько выше нормальных для возраста значений.
Все пациенты с сольтеряющей формой нуждаются в заместительной терапии минералокортикоидами. Единственным препаратом с минералокортикоидными свойствами является флудрокортизон (Кортинефф). Стартовая доза Кортинеффа составляет 0,05–0,2 мг/сут [7]. У новорожденных детей потребность в минералокортикоидах самая высокая – 0,1–0,3 мг/сут. Грудным детям в дополнение к Кортинеффу обычно требуется добавление к пище хлористого натрия (поваренной соли) в виде соленой воды из расчета 1–2 г/сут. В большинстве случаев необходим двукратный прием Кортинеффа в утренние часы (7.00) и в дневное или непозднее вечернее время (15.00–18.00). С возрастом потребность в минералокортикоидах снижается.
Коррекция дозы проводится на основании наличия клинических симптомов дефицита минералокортикоидов: частых срыгиваний, особенно “фонтанирующих”, плохой прибавки в весе, сниженного тургора кожи у маленьких детей, тошноты, плохого аппетита, рвоты, низкого артериального давления у детей более старшего возраста. Для оценки адекватности и решения вопроса о необходимости коррекции дозы Кортинеффа существуют объективные лабораторные критерии: о необходимости увеличения дозы свидетельствуют повышение уровня калия и снижение уровня натрия в крови, рост активности ренина плазмы [5]. Наиболее чувствительным критерием недостаточной дозы Кортинеффа является уровень ренина плазмы, который необходимо измерять всем пациентам вне зависимости от наличия симптомов потери соли. Поскольку повышение уровня ренина предшествует развитию клинических симптомов потери соли, назначение Кортинеффа пациентам с повышенной активностью ренина позволит предотвратить сольтеряющий криз. Кроме того, назначение минералокортикоидных препаратов уменьшает потребность в глюкокортикоидах, следовательно, делает возможным достижение максимального конечного роста [11]. Клиническими симптомами передозировки минералокортикоидов являются отеки, особенно в области лица; повышение АД, беспокойный сон и головные боли. О передозировке и необходимости снижения дозы говорят также тенденция к гипокалиемии и сниженный уровень активности ренина плазмы. Повышение дозы Кортинеффа может потребоваться летом пациентам, проживающим в жарком климате, в связи с тем, что недостаток альдостерона приводит к повышенному выведению натрия через потовые железы.
Наряду с консервативной терапией девочкам с классическими формами АГС проводится хирургическое лечение наружных гениталий. Феминизирующая пластика наружных гениталий выполняется в возрасте 1– лет и включает резекцию клитора с сохранением сосудистонервного пучка, головки и препуциальной кожи, формирование входа во влагалище. При большой степени вирилизации гениталий может потребоваться второй этап оперативного лечения, включающий интроитопластику. Он осуществляется через 1– года после наступления менархе, но до начала половой жизни.
Заключение
АГС –одно из наиболее частых среди наследственных заболеваний эндокринной системы. Своевременная диагностика и адекватное лечение позволяют компенсировать недостаточность функции надпочечников и корректировать отклонения в половом развитии, обеспечивая пациенту высокое качество жизни, фертильность и делая его полноценным членом общества. Введение неонатального скрининга способствует оптимальному решению вопроса своевременной диагностики и лечения.
Информация об авторе:
Карева Мария Андреевна – кандидат медицинских наук, заведующая отделением опухолей эндокриннойсистемы у детей, старший научный сотрудник Института детской эндокринологии ФГУ ЭНЦ.
E-mail: i_marusya@mail.ru

{kind=link}