Введение
Болезнь Помпе (БП), или болезнь накопления гликогена типа II (OMIM 232300), – редкое моногенное заболевание с полисистемными проявлениями. Распространенность заболевания составляет около 1:40 000 до 1:140 000 в зависимости от формы и региона [1]. БП вызывается мутациями в гене, кодирующем синтез фермента кислой α-глюкозидазы (GAA), и имеет аутосомно-рецессивный характер наследования [2]. Ген GAA расположен на длинном плече 17-й хромосомы 17q25.2-25.3.
Проявления БП у детей варьируются от бессимптомного течения с повышением уровня креатининфосфокиназы (КФК) в крови до грубой задержки моторного развития с выраженной мышечной слабостью. Возраст появления симптомов и тяжесть течения зависят от остаточной активности фермента. Выделяют две основные формы заболевания: с ранним (инфантильная форма) и поздним началом. БП с поздним началом подразделяют на детскую (1–5 лет), ювенильную (5–18 лет) и взрослую (старше 18 лет) [3].
Инфантильная форма БП манифестирует в течение первых 6 месяцев жизни прогрессирующей гипертрофией миокарда желудочков, задержкой моторного развития, мышечной гипотонией, гепатомегалией и макроглоссией. В биохимическом анализе крови имеет место повышение содержания внутриклеточных ферментов, таких как КФК, КФК-МВ, аспартатаминотрансферазы (АСТ), аланинаминотрансферазы (АЛТ), лактатдегидрогеназы (ЛДГ) и выраженное повышение уровня натрийуретического пептида (NT-proBNP). Инфантильная форма заболевания носит быстропрогрессирующий характер с развитием тяжелой сердечной и дыхательной недостаточности, что служит причиной летального исхода [4]. Большинство детей умирают в течение первого года жизни в отсутствие своевременного лечения [1, 5].
Стандартом диагностики БП является определение активности фермента α-глюкозиды в сухих пятнах крови с последующим проведением молекулярно-генетического исследования детей с пониженной активностью фермента для выявления мутации в гене GAA [6, 7]. Поскольку БП имеет аутосомно-рецессивный тип наследования, для возникновения заболевания необходимо наличие одной мутации в гомозиготном состоянии или двух различных мутаций в компаунд-гетерозиготном состоянии. Пациентам с двумя различными патогенными вариантами важно подтвердить диагноз с помощью сегрегации родственников, чтобы продемонстрировать, что две мутации находятся в двух разных аллелях.
Важным фактором, влияющим на результат фермент-заместительной терапии (ФЗТ) БП, является определение перекрестно-реактивного иммунологического статуса (CRIM – Cross-reactive immunological material). Принято считать, что у пациентов с инфантильной формой заболевания активность α-глюкозидазы в лейкоцитах составляет менее 1%, т.е. происходит синтез нефункциональной формы фермента, однако у ряда пациентов такой синтез «нативных» ферментов отсутствует полностью. Пациенты с остаточной активностью фермента классифицируются как CRIM-положительные (CRIM+), пациенты с полным отсутствием фермента относятся к CRIM-отрицательным (CRIM-) [8]. Дети с CRIM-отрицательным статусом показывают слабый ответ на проводимую ФЗТ, т.к. их организм распознает рекомбинантную α-глюкозидазу как чужеродный иммуногенный материал и вырабатывает к ней нейтрализующие антитела. Информацию о CRIM-статусе и предполагаемой тяжести течения заболевания при уже известных нуклеотидных вариантах гена GAA можно найти в базе данных мутаций, созданной Центром Помпе в Роттердаме [9]. CRIM-статус определяют методом Вестерн-блоттинга с использованием гомогенатов культивируемых фибробластов либо мононуклеаров периферической крови пациента, однако в настоящее время в России данный анализ не выполняется [10].
Изначально исследования патофизиологии БП были сосредоточены на сердечной и скелетных мышцах, однако в дальнейшем было доказано отложение гликогена в других типах клеток, таких как двигательные нейроны, гладкомышечные и эндотелиальные клетки [11–14]. Гистологическое исследование мышечной ткани для постановки диагноза в настоящее время не является обязательным [15].
Текущий стандарт лечения БП основан на ФЗТ препаратом алглюкозидаза-α – рекомбинантного фермента – предшественника α-глюкозидазы, одобренного в 2006 г.
ФЗТ доступна для всех типов заболевания, но эффекты от нее различны [16–18]. Первоначально рекомендованная доза препарата алглюкозидаза-α составляла 20 мг/кг каждые две недели для всех пациентов с БП. Эта рекомендация основана только на одном исследовании, в котором сравнивались две небольшие группы пациентов с инфантильной формой БП, получавших алглюкозидазу-α в дозе 20 мг/кг/2 нед. либо 40 мг/кг/2 нед., где не было получено разницы между ними в отношении безо-пасности и эффективности режимов дозирования [19]. Следует принять во внимание, что образование антител против α-глюкозидазы ухудшает ответ на ФЗТ и требует дополнительного специфического медикаментозного лечения и необходимости изменения режима дозирования ФЗТ. В последних исследованиях показана эффективность применения более высокой дозы (20–40 мг/кг каждую неделю) препарата пациентами с инфантильной формой, проявляющейся выраженной мышечной слабостью и/или тяжелым течением гипертрофической кардиомиопатии [20–22].
К сожалению, в настоящее время отсутствуют международные рекомендации по лечению инфантильной формы БП, включившие новые знания, полученные в последние годы [23].
Цель исследования: обосновать актуальность определения CRIM-статуса у пациентов с болезнью Помпе и значимость ранней диагностики заболевания.
Методы
Под наблюдением врачей кардиологического отделения ФГАУ НМИЦ здоровья детей Минздрава РФ в период с 2011 по 2020 г. находились 12 пациентов (6 мальчиков и 6 девочек) с инфантильной формой БП, подтвержденной как с помощью энзимодиагностики, так и результатами генетического тестирования, получавших ФЗТ путем внутривенного введения рекомбинантной кислой α-глюкозидазы человека.
В настоящее время два пациента продолжают наблюдение в клинике. Сводные данные по пациентам представлены в табл. 1.
Возраст пациентов на момент постановки диагноза варьировался от 3 до 24 месяцев (М=8 месяцев), ФЗТ была начата в возрасте от 4 до 24 месяцев (М=8 месяцев). При сборе анамнеза были выявлены следующие жалобы: задержка моторного развития, регресс ранее приобретенных моторных навыков (в возрасте от 2 до 6 месяцев, М=4 месяца), одышка, трудности вскармливания, частые респираторные инфекции. При клиническом осмотре у всех детей обращали на себя внимание выраженная мышечная гипотония, задержка моторного развития, периоральный и периорбитальный цианоз, макроглоссия, одышка, гепатомегалия. У трех пациентов отмечены тахикардия и пастозность нижних конечностей, у 1 – спленомегалия и отеки нижних конечностей.
Применены клинические (сбор семейного анамнеза, осмотр пациента); лабораторные (определение уровней КФК, КФК-МВ, ЛДГ, АСТ, АЛТ, натрийуретического пептида); инструментальные (эхокардиография, электрокардиография, рентгенография органов грудной клетки, ультразвуковое исследование почек и органов брюшной полости); молекулярно-генетические (секвенирование гена GAA методом прямого автоматического секвенирования по Сенгеру) и биохимические исследования в объеме энзимодиагностики путем тандемной масс-спектрометрии.
Результаты
При молекулярно-генетическом подтверждении диагноза у восьми детей выявлены мутации в обоих аллелях гена GAA, приводящие к развитию заболевания, у четырех удалось выявить мутацию только в одном из аллелей гена кислой α-глюкозидазы.
У всех детей в биохимическом анализе крови выявлено повышение уровня КФК до 5–6 норм, ЛДГ до 3–4, АЛТ до 5–10, АСТ до 5–10 норм. На фоне проводимой ФЗТ значимой динамики снижения содержания данных ферментов в сыворотке крови не отмечено. У пациентов B, C, D, получавших ФЗТ и наблюдавшихся дольше всего (в течение 12 месяцев), зарегистрированы колебания биохимических показателей без определенной тенденции.
По данным Эхо-КГ, у всех пациентов с инфантильной формой БП выявлена гипертрофия миокарда обоих желудочков. У 7 из них (A, B, C, D, E, F, G) до начала ФЗТ индекс массы миокарда (ИММ) ЛЖ составлял от 233 до 607 г/м2 (Z-score – 7,5–14,3). На фоне длительно проводимой ФЗТ отмечено быстрое уменьшение данного показателя, через 52 недели после начала ФЗТ ИММ ЛЖ составил от 112 до 161 г/м2 (Z-score – 3,7–5,5). Толщина ЗСЛЖ у всех 12 детей до начала ФЗТ составила от 8,5 до 20 мм (Z-score – 3,3–9,58), толщина МЖП от 9 до 21 мм (Z-score – 3,94–13,74). Через 16 недель после начала патогенетической терапии в большинстве случаев отмечено уменьшение выраженности гипертрофии миокарда ЛЖ, у детей, получавших терапию в течение года (n=3), толщина ЗСЛЖ составила от 7 до 12 мм (Z-score – 2,9–6,1), толщина МЖП – 10–12 мм (Z-score – 3,6–4,7). В дебюте заболевания у 7 пациентов сократительная способность ЛЖ была в норме, в 5 случаях отмечено значительное снижение ФВ ЛЖ (от 15 до 36%). К 26-й неделе терапии у всех детей ФВ ЛЖ нормализовалась. Помимо патогенетической всем пациентам была назначена терапия, направленная на коррекцию симптомов ХСН: 10 детей получали петлевые диуретики (A, B, C, D, E, F, G, H, I, J), 7 – ингибиторы АПФ (A, C, D, E, F, G, I), 10 – β-адреноблокаторы (A, B, C, D, E, F, H, I, K, L), 3 – сердечные гликозиды (D, E, G). Также регулярно проводились курсы щадящего периферического массажа.
Ниже представляем два клинических случая с инфантильной формой БП ФГАУ НМИЦ здоровья детей Минздрава РФ и Самарского областного клинического кардиологического диспансера им. В.П. Полякова.
Клинический случай 1
Девочка с CRIM-отрицательным статусом от первой беременности на фоне холестаза, анемии легкой степени тяжести, c тугим обвитием шеи плода пуповиной. Родилась на 39-й неделе гестации естественным путем с массой тела 2690 г, длиной 50 см. Пренатально при ультразвуковом исследовании плода патологии не выявлено, после рождения эхокардиография (Эхо-КГ) не проводилась. При обследовании родителей (Эхо-КГ, электрокардиография – ЭКГ) патологии не выявлено.
Родители отмечали отставание в моторном развитии с 4-месячного возраста. Девочка не удерживала голову, не сидела, отмечались вялость, общая мышечная слабость, стонущее дыхание и сниженный аппетит. Ребенок был госпитализирован в Самарский областной клинический кардиологический диспансер в тяжелом состоянии. При осмотре реакция на болевые раздражители замедлена и снижена, голос тихий, плач непродолжительный, малоэмоциональный, аппетит снижен (из бутылочки сосет вяло и неохотно), одышка с участием вспомогательной мускулатуры с частотой дыхания в покое до 50–65 в минуту, пальпаторно гепатомегалия (+3 см ниже края правой реберной дуги). Обращали внимание диффузная мышечная гипотония, макроглоссия и гиперсаливация.
По результатам лабораторного обследования: резкое повышение уровней АСТ (317,7 ЕД/л при норме до 35,0 ЕД/л), АЛТ (217,8 ЕД/л при норме до 40,0 ЕД/л), КФК (1425 ЕД/л при норме до 172,0 ЕД/л), КФК-МВ (62,5 ЕД/л при норме до 24,0 ЕД/л), ЛДГ (2189 ЕД/л при норме до 248,0 ЕД/л), NT-proBNP (12891 пг/мл при норме до 125,0 пг/мл), высокочувствительного тропонина (0,172 нг/мл при норме до 0,02 пг/мл).
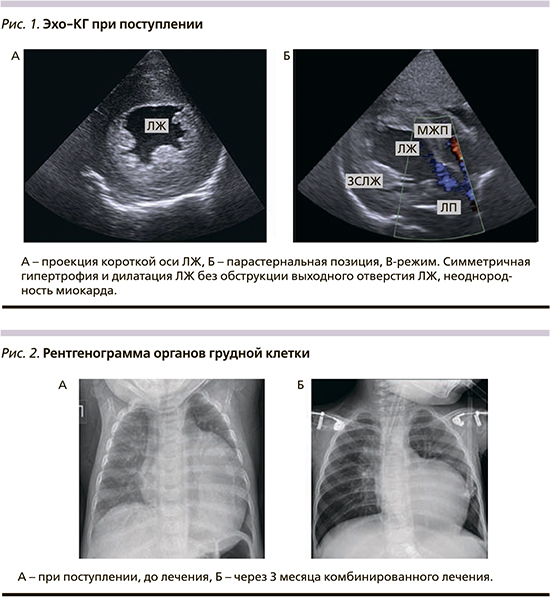
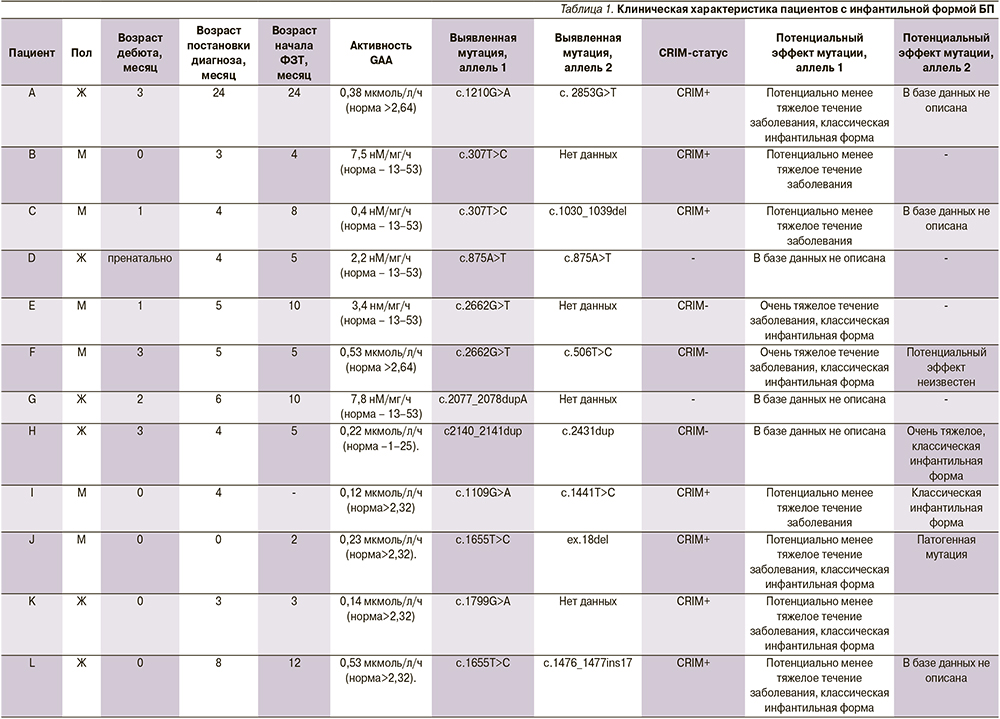
По Эхо-КГ (рис. 1) резко увеличен объем полости левого желудочка (ЛЖ): конечный диастолический размер – КДР ЛЖ 39 мм (Z-score +5), гипертрофия стенок ЛЖ – (межжелудочковая перегородка – МЖП 12 мм (Z-score +6,59), задняя стенка ЛЖ 11 мм (Z-score +4,6)), индекс массы миокарда левого желудочка 432 г/м², снижена сократительная способность миокарда ЛЖ (фракция выброса 31% Simpson), недостаточность на атриовентрикулярных клапанах 2-й степени, повышение давления в системе легочной артерии (42 мм рт.ст.). На рентгенограмме органов грудной клетки (рис. 2) выявлена кардиомегалия (кардиоторакальный индекс 0,7). На ЭКГ – ускорение атриовентрикулярной проводимости с признаками неполной блокады правой ножки пучка Гиса, вольтажные признаки гипертрофии обоих желудочков, выраженные изменения трофики миокарда и признаки субэндокардиальной ишемии.
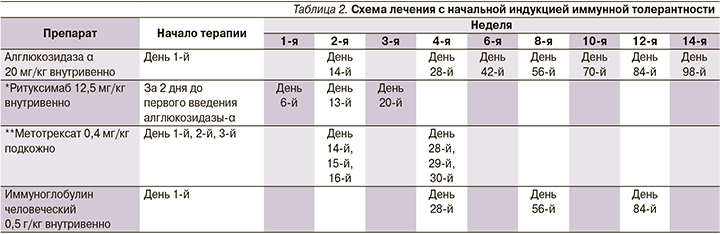
Учитывая сочетание кардиомегалии, гипертрофии миокарда с мышечной гипотонией, макроглоссией, лабораторные изменения, заподозрен диагноз «болезнь Помпе». По результатам энзимодиагностики выявлено резкое снижение активности α-глюкозидазы (0,06 мкмоль/л/ч при норме от 1,0 до 25,0 мкмоль/л/ч), по результатам секвенирования гена GAA выявлены ранее описанные нуклеотидные варианты: c.118C>T, приводящий к замене p.Arg40*, и c.2662G>T, приводящий к замене p.Glu888*, в компаунд-гетерозиготном состоянии. При анализе выявленных мутаций в соответствии с базой данных Pompe disease GAA variant database отмечена ассоциация с потенциально очень тяжелым течением заболевания, оба нуклеотидных варианта имели CRIM-отрицательный статус [9, 24]. Учитывая CRIM-отрицательный статус пациента, принято решение предварительно провести индукцию иммунной толерантности. Наиболее изученной и эффективной является схема с комбинацией, включающей 3 препарата: ритуксимаб, метотрексат, внутривенные иммуноглобулины (табл. 2) [3, 25, 26]. С целью купирования симптомов хронической сердечной недостаточности (ХСН) ребенок получал комбинированную терапию в соответствии с клиническими рекомендациями: диуретик, ингибитор ангиотензинпревращающего фермента (АПФ), β-адреноблокатор, антагонист минералкортикоидных рецепторов. Учитывая тяжелое состояние ребенка при поступлении, не удалось избежать подключения искусственной вентиляции легких (ИВЛ).
При оценке состояния пациентки через 3 месяца от начала лечения отмечено улучшение эмоционального фона, она начала улыбаться, появилась мышечная сила в руках, однако продолжалась аппаратная вентиляции через трахеостому с умеренными параметрами. По данным лабораторных исследований отмечено снижение уровня КФК с 1425 до 330,8 ЕД/л, КФК-МВ с 62,5 до 27,9 ЕД/л, ЛДГ с 2189 до 1370,5 ЕД/л, NT-proBNP с 12891 до 1810 пг/мл, высокочувствительный тропонин I c 0,172 до 0,03 нг/мл, АСТ с 317,7 до 174,7 ЕД/л. Согласно результатам Эхо-КГ уменьшился КДР ЛЖ с 39 до 31 мм (Z-score +2,16), увеличилась фракция выброса ЛЖ (с 31 до 58%), уменьшился индекс массы миокарда ЛЖ с 432,0 до 323,7 г/м2. На рентгенограмме органов грудной клетки уменьшился кардиоторакальный индекс (КТИ) с 0,7 до 0,58.
Молекулярно-генетическое обследование матери показало наличие нуклеотидного варианта c.118C>T, приводящего к замене p.Arg40* в гетерозиготном состоянии; нуклеотидного варианта c.2662G>T, приводящего к замене p.Glu888* не выявлено. Молекулярно-генетическое обследование отца ребенка не проводилось (отказался от обследования).
Этот клинический случай демонстрирует эффективность предварительно проведенной индукции иммунной толерантности у пациента с CRIM-отрицательным статусом, без проведения иммуносупрессии, вероятно, быстрое формирование антител к алглюкозидазе α, что, безусловно, снизило бы эффективность проводимой ФЗТ.
Клинический случай 2
Мальчик 3,5 года с CRIM-положительным статусом наблюдается в кардиологическом отделении ФГАУ НМИЦ здоровья детей Минздрава РФ с возраста 1 месяц. Ребенок от 2-й беременности, протекавшей на фоне угрозы прерывания на 22-й неделе гестации, острой респираторной вирусной инфекции на 24-й и 37-й неделях гестации. Роды вторые, оперативные, в связи с угрозой несостоятельности рубца на матке. Масса тела при рождении 3500 г, длина 54 см. Оценка по шкале Апгар 8/9 баллов. Семейный анамнез отягощен: у сибса мужского рода летальный исход в возрасте 1 года 3 месяцев в связи с поздно диагностированной БП. ФЗТ не проводилась.
После рождения состояние ребенка тяжелое за счет судорожного синдрома. По результатам лабораторно-инструментального обследования выявлены высокие показатели КФК 1668 ЕД/л, АСТ 279 ЕД/л, АЛТ 69 ЕД/л, ЛДГ 1705 ЕД/л. По данным Эхо-КГ, проведенной на 7-е сутки жизни, отмечено умеренное расширение правого предсердия, утолщение МЖП до 4,5 мм, открытое овальное окно 4 мм с лево-правым сбросом. Принимая во внимание сочетание отягощенного семейного анамнеза, гипертрофии миокарда ЛЖ и лабораторные данные, проведена энзимодиагностика БП методом транскраниальной магнитной стимуляции, при которой выявлено снижение активности α-глюкозидазы (0,12 мкмоль/л/час при норме >2,32 мкмоль/л/час).
Молекулярно-генетический анализ: в экзоне 13 гена GAA выявлена мутация c.1655T>C, приводящая к аминокислотной замене p.Leu552Pro, описана в базе данных как ассоциированная с классической инфантильной формой заболевания, имеет CRIM-положительный статус, а также делеция в 18 экзоне гена GAA ex.18del в компаунд-гетерозиготном состоянии. Молекулярно-генетическое обследование родителей пробанда (ген GAA): у мамы мутация с.1655Т>C в гетерозиготном состоянии, у отца – ex.18del в гетерозиготном состоянии.
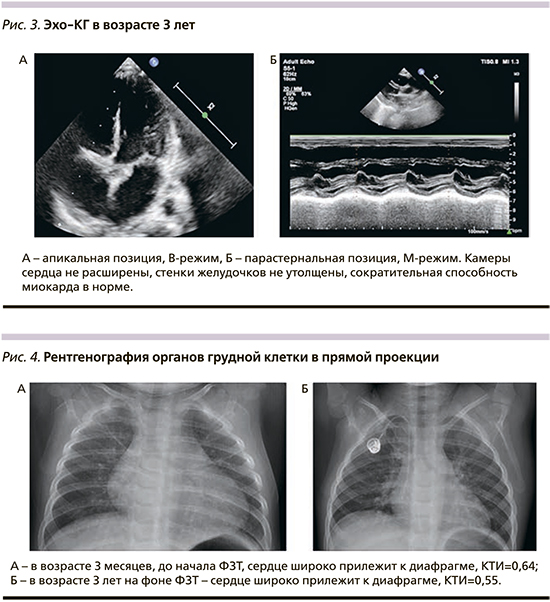
С возраста 3 месяцев получает ФЗТ препаратом алглюкозидаза α из расчета 20 мг/кг/2 нед. Поскольку на основании генетического анализа пациент имел CRIM-положительный статус, ФЗТ инициирована без применения индукции иммунной толерантности. При обследовании на фоне лечения в возрасте 3 лет: контакту доступен, понимает обращенную речь, выполняет инструкцию, навыки опрятности и самообслуживания в стадии формирования; речь фразовая, дизатричная, периодически заикание, птоза, нистагма нет, лицо симметрично, гипомимично, глотание не нарушено, глоточные рефлексы снижены, гиперсаливация, дисфония; объем активных и пассивных движений не нарушен, мышечный тонус диффузно снижен, сила мышц в дистальных отделах снижена до 4 баллов, в проксимальных до 4–3 баллов, сухожильные рефлексы с рук средней живости, с ног значительно снижены; патологические рефлексы не вызываются; моторные навыки: сидит, садится, стоит, встает, используя миопатические приемы, ходит, бегает, но без отрывания ног от поверхности пола. По данным лабораторного обследования сохраняются повышение уровней АСТ (286,69 при норме <42 ЕД/л), АЛТ (169,63 при норме <40 ЕД/л), ЛДГ (833,68 при норме 91,0–295,0 ЕД/л), NT-proBNP (193,5 при норме <62 пг/мл). Отмечена положительная динамика параметров Эхо-КГ: полости сердца не расширены, сократительная способность не снижена, повышенная трабекулярность апикального и медиального сегментов ЛЖ, диастолическая функция ЛЖ не нарушена, межжелудочковая перегородка – 4,7 мм (в медиальном отделе 5,5 мм, Z-score – 1,21), задняя стенка ЛЖ – 4,5 мм (не утолщена, Z-score – 0,29), недостаточность митрального клапана +1, недостаточность трикуспидального клапана +1,5 (рис. 3). Рентгенологическая картина в возрасте 3 месяцев и 3 лет также показала положительную динамику в виде уменьшения размера кардиоторального индекса и представлена на рис. 4. В комплексе с ФЗТ с возраста 1 месяца получает β-адреноблокатор и антагонист минералокортикоидных рецепторов.
Второй клинический случай демонстрирует эффективность применения ФЗТ при своевременной диагностике заболевания и инициации лечения. Принимая во внимание CRIM-положительный статус пациента, предварительной индукции иммунотолерантности не проводилось, при дальнейшем наблюдении показано регулярное исследование уровня антител к алглюкозидазе-α.
Обсуждение
Инфантильная форма БП дебютирует при рождении или в течение первых месяцев жизни, ее симптомами являются тяжелая задержка или регресс моторного развития, гепатомегалия, макроглоссия, гипертрофическая кардиомиопатия и кардиореспираторная недостаточность. Без своевременного начала ФЗТ летальный исход наступает на первом году жизни [27].
В 2006 г. FDA (Food and Drug Administration) одобрило алглюкозидазу-α для лечения БП. Доступность ФЗТ изменила естественное течение БП, значительно увеличив выживаемость и улучшив долгосрочные клинические результаты. Несмотря на возможности ФЗТ, общий ответ неоднороден и зависит от множества факторов, включая возраст пациента на момент инициации ФЗТ, степень выраженности ХСН и фенотипа ремоделирования сердца, дозу ФЗТ и выработку антител в ответ на введение алглюкозидазы-α [25], ограниченное поглощение фермента в мышцах, неспособность препарата преодолевать гематоэнцефалический барьер и вариабельность реакции скелетных мышц [28, 29].
На фоне проводимой ФЗТ у части пациентов в нашей когорте отмечена положительная динамика в течении заболевания: улучшился эмоциональный статус, появились новые моторные навыки, купированы проявления ХСН. Результаты представлены в табл. 3.
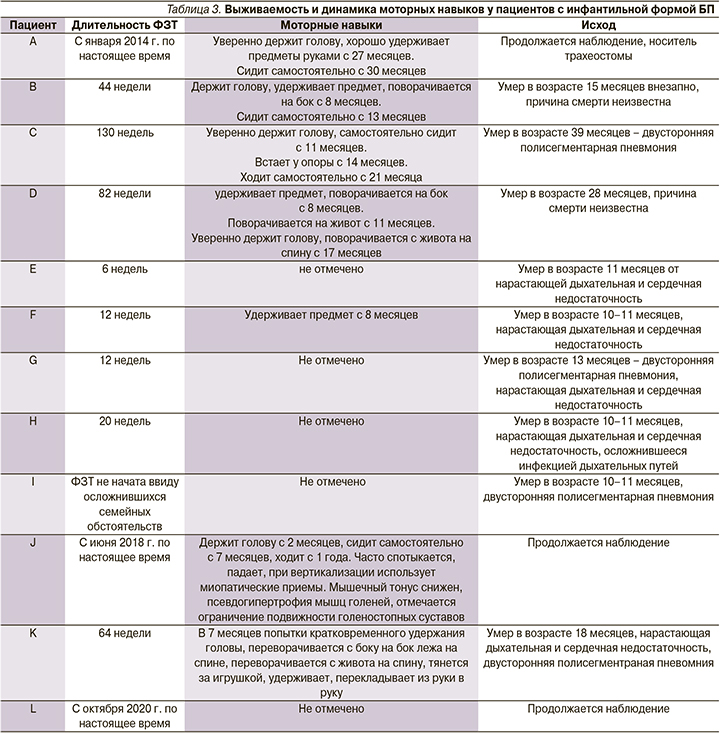
У 4 пациентов (E и G) не наблюдалось положительной динамики в течении заболевания, что наиболее вероятно связано с поздним началом патогенетической терапии, летальный исход наступил вследствие нарастающей дыхательной и сердечной недостаточности. Несмотря на первоначально обнадеживающие результаты лечения, 3 детей (В, С, D) умерли после 44–130 недель ФЗТ: 2 пациента умерли внезапно, у пациента С развилась тяжелая дыхательная и сердечная недостаточность на фоне инфекционного заболевания, приведшая к летальному исходу. Наиболее эффективна ФЗТ для пациента J (клинический случай 2) с началом лечения практически сразу после рождения до появления клинической картины заболевания: у мальчика отмечена умеренная мышечная гипотония, приобретение моторных навыков в соответствии с возрастными нормами, имеет место задержка речевого развития, обусловленная мышечной слабостью. Безусловно, поздняя диагностика БП, соответственно, позднее начало патогенетического лечения определяют неблагоприятный исход заболевания ввиду развития необратимых изменений в мышечной ткани.
Исследование 2010 г., оценивавшее влияние CRIM-статуса на результаты лечения БП, показало, что пациенты с CRIM-отрицательным статусом, получавшие монотерапию ФЗТ, либо умерли, либо зависели от аппарата ИВЛ в возрасте 27,1 месяца из-за выработки антител к вводимой алглюкозидазе-α [8]. Было показано, что почти у всех CRIM-отрицательных и некоторых CRIM-положительных пациентов развиваются высокие титры нейтрализующих антител при ФЗТ. Чтобы избежать образования высоких титров антител у CRIM-отрицательных пациентов, была предложена профилактическая индукция иммунной толерантности с помощью ритуксимаба, метотрексата и внутривенных иммуноглобулинов (ВВИГ) [24, 26]. Этот подход также был применен к CRIM-положительным субъектам, у которых развиваются высокие титры антител [24, 26]. Быстрое определение CRIM-статуса важно и должно быть проведено до инициации ФЗТ, но не должно задерживать начало ФЗТ. Следовательно, пациентов с инфантильной формой БП с неизвестным CRIM-статусом следует рассматривать как CRIM-отрицательных. Схема медикаментозной индукции иммунной толерантности для пациентов с инфантильной формой БП впервые разработана для уменьшения развития и минимизации влияния антител к α-глюкозидазе на терапевтический ответ на ФЗТ и стала стандартом лечения CRIM-отрицательных пациентов с инфантильной формой БП [24, 26, 30–32]. К сожалению, при лечении большинства наших пациентов мы не имели возможности исследовать CRIM-статус и проводить профилактическую индукцию иммунной толерантности, что отчасти помимо позднего начала может обусловливать неблагоприятные исходы болезни.
В настоящее время продолжаются исследования по выработке оптимального алгоритма проведения ФЗТ у пациентов с БП с недостаточным клиническим эффектом при использовании рекомендованной дозы и кратности введения алглюкозидазы-α.
Исследования E. Poelman et al. (2020) показали, что пациенты, получавшие более высокие дозы ФЗТ (еженедельное введение препарата в дозе 20–40 мг/кг либо 40 мг/кг/2 нед.), имели лучшую динамику по моторному развитию, респираторной функции и показателям биохимических маркеров [33]. Также в статье, опубликованной группой авторов из США, продемонстрировано существенное улучшение моторных навыков и функции дыхания при увеличении дозировки ФЗТ до 40 мг/кг/нед. по сравнению с использованием стандартной дозы 20 мг/кг/2 нед. [21].
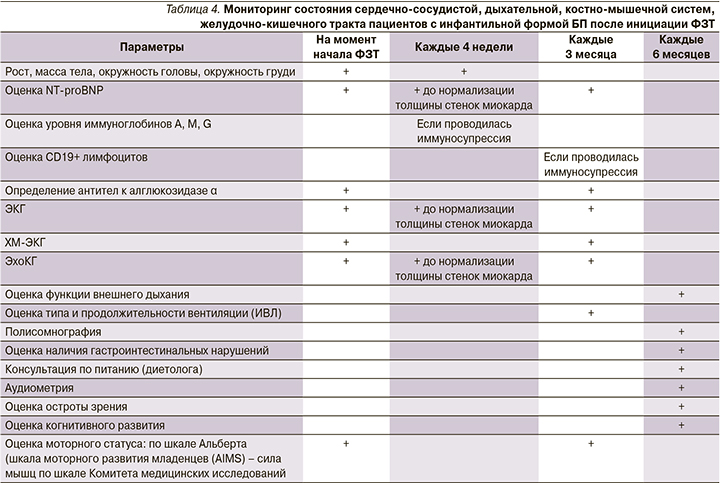
Высокая выживаемость пациентов с инфантильной формой заболевания привела к новым фенотипам с сердечными, речевыми, слуховыми, скелетно-мышечными, респираторными, глотательными и нейрокогнитивными нарушениями. В опубликованной литературе продемонстрировано, что у пациентов в отдаленные сроки наблюдаются мышечная слабость, речь с «гнусавым» оттенком, дисфагия с риском аспирации, птоз и аритмия [34]. Пациентам с инфантильной формой БП после инициации ФЗТ рекомендован регулярный мониторинг клинического состояния и лабораторно-инструментальных параметров (табл. 4) [35].
Заключение
За последние 15 лет благодаря ФЗТ появилась возможность оказания медицинской помощи детям с инфантильной формой БП, однако в то же время заболевание из быстропрогрессирующего и фатального на первом году жизни превратилось в хроническое все еще с высокой летальностью. Наиболее перспективна ФЗТ при ее начале до появления клинических проявлений, что делает крайне важной раннюю диагностику заболевания. Индукция иммунной толерантности у CRIM-негативных пациентов может позволить уменьшить образование антител к алглюкозидазе-α, позволив усилить эффективность терапии детей данной категории. У пациентов с БП, демонстрирующих недостаточный клинический ответ на стандартные схемы ФЗТ, может быть рассмотрено повышение дозы и/или кратности введения препарата. Поскольку в настоящее время нет единого алгоритма ведения пациентов с инфантильной формой, необходимы дальнейшие исследования для улучшения качества оказываемой помощи.
Генная терапия с использованием векторов, нацеленных не только на скелетные и сердечные мышцы, является дальнейшим перспективным направлением исследований [23].
Финансирование. Отсутствует.
