Обоснование
Субкорнеальный пустулезный дерматоз (СПД) – это редкое хроническое рецидивирующее заболевание с симметричным пустулезным поражением туловища, крупных складок и преимущественно сгибательных участков конечностей. Бывает идиопатическим или ассоциированным с онкологическими, аутоиммунными заболеваниями (например, с болезнью Крона) или инфекциями [1]. Относится к группе нейтрофильных дерматозов – состояний, при которых обнаруживается воспалительный инфильтрат из зрелых полиморфноядерных лейкоцитов [2]. По данным литературы, наиболее типичен для женщин средней возрастной группы, описаны единичные случаи начала в детском возрасте [3].
Препаратом выбора терапии нейтрофильных дерматозов служит дапсон, но в некоторых случаях могут оказаться эффективными системные глюкокортикостероиды (при иммуноглобулин-А (IgA)-позитивном варианте, который рассматривается сегодня как подтип пузырчатки) [4]. IgA-пузырчатка, гистологически и клинически не отличимая от СПД, относится к одному из подтипов (СПД-тип) аутоиммунного буллезного дерматоза группы пузырчатки, при котором выявляются аутоантитела класса А к десмоколлину-1 [5]. По данным M. Tajima et al., описано немногим более 70 случаев [6], при этом возраст начала варьировался от 1 месяца до 85 лет, также с предоминированием у женщин средней возрастной группы [7]. Решающими в случае дифференциальной диагностики считаются данные, полученные с использованием дополнительных методов лабораторного исследования [8].
Клинический случай
В кожное отделение СПбГПМУ 15.07.2019 была госпитализирована девочка Ф. 17 лет 10 месяцев, которая остро заболела 08.07.2019, когда впервые появились высыпания на шее и передней поверхности грудной клетки, в последующем распространившиеся на волосистую часть головы и конечности. Известно, что за несколько дней до высыпаний отмечались симптомы поражения респираторного тракта (ринорея, субфебрилитет, кашель), которые купировались благодаря симптоматической терапии. Из анамнеза жизни стало известно, что девочка рождена от 3-й беременности, протекавшей на фоне гестоза и угрозы прерывания в ходе 3 родов на 34-й неделе гестации от матери с отрицательным Rh-статусом. Длительное время после рождения Ф. провела в отделении интенсивной терапии, о чем документальные свидетельства предоставлены не были. С момента появления на свет ребенок находился на искусственном вскармливании и под наблюдением невролога с диагнозом «детский церебральный паралич. Органическое поражение центральной нервной системы. Симптоматическая эпилепсия». Из перенесенных детских инфекционных заболеваний в анамнезе только ветряная оспа, прививки не были проведены в связи с наличием противопоказаний. В семейном анамнезе кожные заболевания отсутствовали. Ранее у девочки также не отмечалось признаков хронических дерматозов, как и проявлений аллергических реакций. При поступлении состояние было расценено как средней степени тяжести. Общение с девочкой было крайне затруднено, т.к. речь отсутствовала, ребенок общался звуками, периодически демонстрируя немотивированное возбуждение, сопровождавшееся вокализацией и нистагмом. Пациентка самостоятельно не сидела, не передвигалась, не была способна поддерживать позу. При осмотре отмечалась выраженная гипотрофия, рост – 112 см, масса тела – 12,5 кг, индекс массы тела – 9,9 (дефицит массы тела по росту – 34%), все показатели были ниже 5-го перцентиля по шкале GMFSC V (Gross motor function classification system), что соответствует тяжелой белково-калорической недостаточности. Твердую пищу ребенок не употреблял, только жидкую или в перетертом виде. При физикальном осмотре была выявлена гепатомегалия (+2 см от реберного края), пальпации были доступны облигатные группы лимфатических узлов. Поражение кожи на момент поступления носило распространенный характер в виде мелких пустул на эритематозном фоне, склонных к слиянию с образованием нумулярных бляшек с серозно-геморрагическими крупными корками на поверхности неправильной формы или кольцевидными, гирляндообразными элементами, располагавшимися на шее, туловище, в аксиллярных и паховых областях, частично с переходом на разгибательные поверхности конечностей, преимущественно верхних, более слева с захватом тыла кисти и двух дистальных фаланг 3-го и 4-го пальцев кисти без изменения ногтевых пластин (рис. 1, 2).
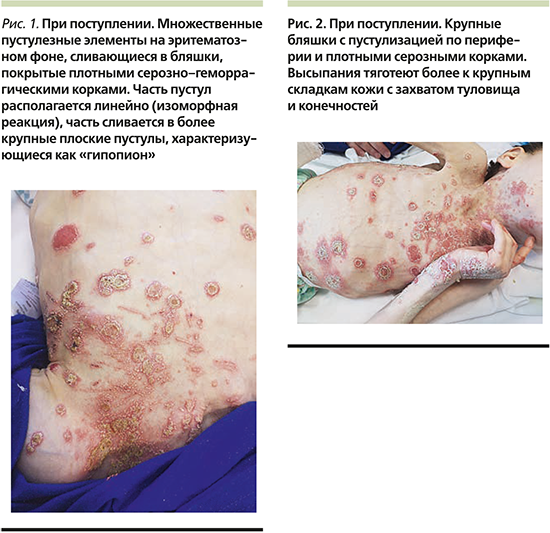
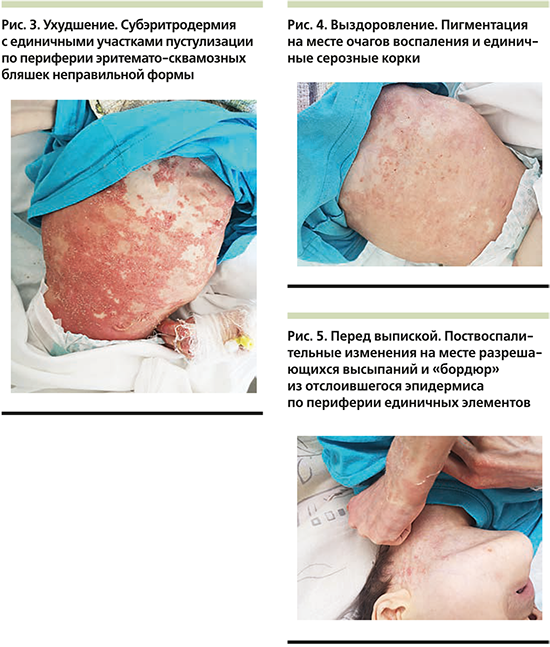
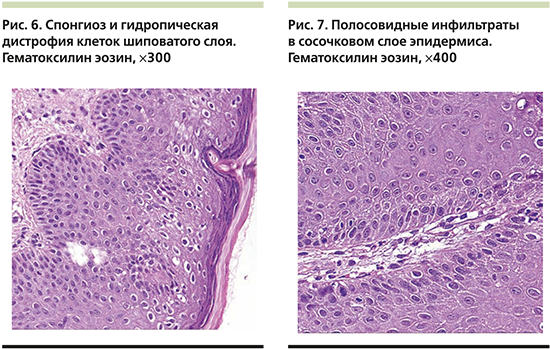
На волосистой части головы отмечались единичные бляшки до 3 см в диаметре, покрытые плотными слоистыми чешуйко-корками с пустулизацией по периферии. Часть пустул располагалась линейно в местах механической травматизации по типу изоморфной реакции. Не представлялось возможным оценить наличие зуда. Лабораторные показатели были значительно изменены: так, отмечался лейкоцитоз до 14,1×109/л с выраженным сдвигом влево вплоть до юных форм c умеренной токсической зернистостью гранулоцитов, увеличением скорости оседания эритроцитов (СОЭ) до 34 мм/ч, повышением уровня трансаминаз: аланинаминотрансфераза (АЛТ) – 136 (0–40) ЕД/л, аспартатаминотрансфераза (АСТ) – 223 ЕД/л, и С-реактивного белка (СРБ) – 33 (0–5) мг/л, а также гипоальбуминемия – 27,96 (32–45) г/л. Обследование на хронические инфекционные заболевания выявило наличие IgG к Mycoplasma pneumoniaе и вирусу Эпштейна–Барр, при этом общие показатели иммуноглобулинов различных классов (иммунология первого порядка) соответствовали возрастной норме. Девочка получала инфузионную терапию глюкозо-солевыми растворами, антибактериальную терапию (цефоперазон и сульбактам), противогрибковую терапию (флуконазол), системные глюкокортикостероиды из расчета 2 мг/кг массы тела, противоэпилептическую терапию (вальпроевая кислота, карбамазепин), пробиотическую терапию (бифидо- и лактофлора), заместительную терапию (панкреатин, урсодезоксихолевая кислота), дополнительные питательные смеси и витамины (Пептамен Юниор, витами D). На третьи сутки от момента поступления состояние пациентки ухудшилось (поражение кожи распространилось более чем на 60% площади поверхности тела в виде обилия пустул на эритематозном фоне с тенденцией к периферическому расположению, гирляндообразно, кольцевидно (рис. 3), отмечалась судорожная готовность, была зарегистрирована гипокальциемия до 1,1 ммоль/л на фоне дефицита витамина D (4,1[25–80] нг/мл) и вторичного гиперпаратиреоза – паратиреоидный гормон 323,9 пг/мл (12,0–95,0, кровь была взята экстренно после инфузии 200 мл физиологического раствора), нарастание числа лейкоцитоза до 20×109/л (гранулоцитарного: палочкоядерные – 19%, сегментоядерные – 67%, CРБ до 47 мг/л, АСТ до 280 ЕД/л, тропонина – 82,4 (0–17,5) пг/мл, креатинфосфокиназы – 877 ЕД/л [менее 167 мл/л], лактатдегидрогеназы – 1276 ЕД/л, γ-глутамилтранспептидазы – 477 ЕД/л [менее 33 ЕД/л]), несмотря на проведенную терапию, что обусловило перевод девочки в отделение интенсивной терапии для обеспечения адекватного контроля основных показателей жизнедеятельности. Был проведен совместный осмотр в составе педиатра, невролога, гастроэнтеролога, эндокринолога, дерматовенеролога и клинического фармаколога, на котором постановили, что общая тяжесть состояния пациентки обусловлена значительными электролитными нарушениями и выраженным воспалительным процессом, к которому привели токсический гепатит, разившийся на фоне белково-калорической недостаточности, дефицит основных витаминов и микроэлементов, длительный прием противоэпилептических препаратов и предшествовавшее инфекционное заболевание, которое спровоцировало высыпания на коже и нарушение функций внутренних органов. В палате интенсивной терапии девочка провела 3 дня, где дополнительно получала парентеральное питание, были скорректированы объем инфузионной терапии, а также дозы противосудорожной группы по результатам исследования вальпроевой кислоты в крови пациентки, были добавлены эссенциальные фосфолипиды. Наружная обработка все это время была направлена на профилактику вторичного инфицирования кожных покровов (антисептики) и уменьшение воспалительного процесса в коже (топические глюкокортикостероиды). Дальнейшее лечение продолжилось после стабилизации состояния (уменьшение судорожной активности, улучшение биохимических показателей крови: общий кальций – 4,04 ммоль/л, АЛТ – 100 ЕД/л, АСТ – 117 ЕД/л, альбумин – 40,5 г/л) в кожном отделении с привлечением диетолога, который подробно скорректировал суточный рацион питания девочки. Улучшения общего состояния, а также основных биохимических показателей удалось добиться к 02.08.2019, кроме того, произошел регресс кожных высыпаний: отмечались единичные крустозные очаги и поствоспалительная пигментация (рис. 4, 5), с чем пациентка была выписана под наблюдение и проведения дальнейшей терапии при участии педиатра, невролога, дерматовенеролога, эндокринолога по месту жительства и подробными рекомендациями по уходу.
Дифференциальный диагноз
Этиопатогенез СПД остается неизвестным, но описанные в медицинской литературе случаи были связаны с моноклональными гаммапатиями, такими как IgA и IgG, множественной миеломой, хроническим лимфолейкозом, некоторыми сόлидными опухолями (метастатическая тимома, эпидермоидная карцинома легких), отмечалась связь развития дерматоза на фоне поражения респираторного тракта M. pneumoniaе [9, 10]. В случае с паценткой Ф. высыпания на коже появились спустя 2 недели после перенесенного респираторного заболевания, вызванного M. pneumoniaе. Основными для дифференциальной диагностики предполагаются следующие заболевания: IgA-пузырчатка, листовидная пузырчатка, варианты пустулезного псориаза (генерализованный и аннулярный), острый генерализованный экзантематозный пустулез, бактериальное импетиго, дерматофитии, герпетиформный дерматит, некролитическая мигрирующая эритема и др. [11, 12]. Диагноз устанавливался на основании клинической картины и результатов гистологического исследования пораженной кожи с привлечением дополнительных методов окраски, с использованием иммуногистохимических маркеров. Трудности вызывали сходство клинической картины пустулезного псориаза (аннулярный вариант наиболее часто встречается в педиатрической практике) и IgA-пузырчатки. Для верификации последней необходимо обнаружение антител класса IgA к десмосомальным кадгеринам (десмоколлину1–3, и/или десмоглеинам 1, 3) методами твердофазного иммуноферментного анализа или непрямой иммунофлуоресценции [13, 14]. В нашем случае клинические симптомы и патоморфологические признаки (представлены в описании к рис. 6–8) соответствовали таковым при СПД. Возможность проведения иммуногистохимического исследования биоптата на наличие экспрессии иммуноглобулинов с пораженных участках позволила более точно установить подтип СПД (рис. 9–10), но подтвердить серологически наличие аутоантител к структурам десмосом нам не удалось, т.к. в нашем распоряжении не было на том этапе специфических тест-систем. Кроме того, при ухудшении состояния у пациентов с дерматозами нейтрофильной группы описаны общие симптомы, мониторинг которых требует дополнительных лабораторных исследований: развернутого общего анализа крови для выявления лейкоцитоза, биохимического определения электролитов (гипокальциемия) и трансаминаз (гепатит) [15, 16]. Трудности в верификации этиопатогенеза подобных лабораторных признаков в случае с Ф. вызывал общий статус пациентки с тяжелой белково-калорической недостаточностью и явлениями гепатита уже на этапе поступления, которые могли не иметь взаимосвязи с развивающимся нейтрофильным воспалением в коже. Девочка получала системную глюкокортикостероидную терапию, как и рекомендовано в лечении дерматозов группы пузырчатки, а также при некоторых торпидных к сульфонам нейтрофильных пустулезах с постепенным снижением дозы после достижения клинического эффекта [17].
В дальнейшем мама Ф. не обращалась к нам, т.к. ребенок был выписан с исчерпывающими рекомендациями по уходу и дальнейшему лечению в удовлетворительном состоянии на этапе перехода под наблюдение специалистов вне педиатрической сети. Прогноз в случае IgA-пузырчатки, по данным литературы, благоприятный с длительным периодом ремиссии при достижении полного очищения кожи после первого эпизода [7], но прогнозировать что-либо при таком соматическом фоне, как у пациентки Ф., крайне затруднительно.
Обсуждение
СПД также известен как болезнь Снеддона–Уилкинсона. Это редкое хроническое заболевание кожи, характеризующееся образованием под роговым слоем эпидермиса пустул. Впервые заболевание было описано в 1956 г. И.Б. Снеддоном (Ian Sneddon) и Д.С. Уилкинсоном (Darrell Wilkinson). Классическая форма получила название «субкорнеальный пустулез Снеддона–Уилкинсона» [18].
Диагноз устанавливается на основании клинических и патоморфологических признаков, также неизменных с 1956 г. Возникает внезапно в виде поверхностных зудящих или болезненных папул, которые трасформируются в плоские пустулы, везикулы, редко буллы. В течение нескольких суток появляются свежие элементы и образуются кольцевидные, дуговидные, серпигинозные паттерны на коже с центральным западением и пустулами по периферии. Основная часть элементов появляется на неизмененной коже, но в основании некоторых пустул может быть эритематозная или инфильтрированная кожа. Содержимое пустулы разделяется на две субстанции: нижняя часть – гнойная, верхняя – серозная, что обозначают как «гипопион». Пустулы спонтанно вскрываются, содержимое ссыхается в корки, после отпадения которых остаются участки гиперпигментации. Элементы на разных стадиях развития располагаются симметрично, но более сконцентрированы во всех крупных складках, а также на туловище и проксимальной части конечностей. Ногтевые пластинки, ладони и подошвы редко вовлечены в процесс. В большинстве случаев общая симптоматика отсутствует, но имеются данные о возможности их проявления в виде слабости, лихорадки, артралгий, повышения уровня трансаминаз и асептических нейтрофильных абсцессов или склерозирующего гломерулонефрита.
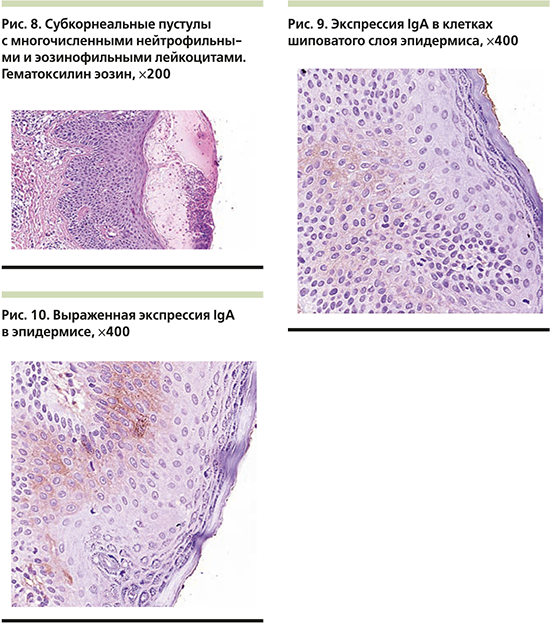
При микроскопическом исследовании субкорнеальный пустулез характеризуется образованием пустул, расположенных непосредственно под роговым слоем эпидермиса, содержащих нейтрофильные и немногочисленные эозинофильные лейкоциты, фибрин, эпителиоциты, иногда немногочисленные лимфоциты (рис. 8). Кроме того, в эпидермисе отмечаются умеренно выраженный акантоз, спонгиоз, итрацеллюлярный отек в шиповатом слое и вторичный акантолиз (рис. 6).
В сосочковом слое дермы определяется инфильтрация клетками, аналогичными составу пустул. Эти инфильтраты линейные, направлены перпендикулярно базальной мембране эпидермиса, местами разрушают базальный слой, проникая в виде единичных клеток в более поверхностные слои многослойного плоского ороговевающего эпителия (рис. 8), вплоть до рогового слоя, образуя описанные выше пустулы. Сосуды сосочкового слоя дермы резко растянуты вследствие выраженного полнокровия, вокруг них наблюдаются мелкоочаговые периваскулярные, преимущественно лимфогистиоцитарные инфильтраты. Выраженность всех гистологических изменений может разниться в зависимости от времени существования взятого для исследования элемента.
При проведении иммуногистохимического исследования СПД можно подразделить на две формы: IgA-негативный и IgA-позитивный [19, 20].
IgA-негативный СПД не имеет каких-либо иммуногистохимических маркеров. Однако при IgA-позитивной форме отмечается положительная экспрессия IgA в поверхностных слоях эпидермиса. Распределение и выраженность положительной реакции несколько неравномерные, в ряде случаев наблюдается очаговое накопление.
Кроме того, при проведении периодического иммуногистохимического исследования пациентов с IgA-негативным вариантом СПД в течение нескольких лет наблюдается прогрессирование в IgA-позитивную форму.
Первыми случаи с клиническим проявлением как при СПД и интерцеллюлярными депозитами IgA описали D. Wallach et al. [9]. Ранее присутствие эпидермального IgA упоминалось G.A. Varigos, I.B. Sneddon и D.S. Wilkinson [21]. Следом за D. Wallach et al. сообщения о возрастающем числе случаев с интраэпидермальными IgA-депозитами стали описывать под различными наименованиями: интраэпидермальный IgA-пустулез [19], атипичный нейтрофильный дерматоз с субкорнеальными IgA-депозитами [20], интерцеллюлярный IgA-дерматоз [22], IgA-пузырчатку или IgA-листовидную пузырчатку, интраэпидермальный нейтрофильный IgA-дерматоз [23], интерцеллюлярный IgA-везикулопустулезный дерматоз [24].
Открытым остается вопрос: принадлежат ли состояния с интраэпидермальными IgA-отложениями к группе аутоиммунных заболеваний, обусловленных аутоантителами класса IgA [25, 26] или они могут быть интегрированы в группу поверхностных нейтрофильных дерматозов? Возможен вариант, предложенный в 1992 г. D. Wallach, считать интраэпидермальный IgA-пустулез частью группы нейтрофильных дерматозов с преувеличенным иммунным ответом. Весьма вероятно, что у части пациентов имеются специфические IgA-аутоантитела к мишеням эпидермальных антигенов, которые приводят к повреждению тканей и воспалительной реакции [27]. Есть предположение относить их к группе больных интерцеллюлярным IgA-дерматозом [28] с шестью различными вариантами, основанными на клинических, патоморфологических и иммунопатоморфологических критериях [29, 30].
Так как СПД – редкое заболевание, сложно организовать и провести рандомизированные контролируемые исследования для определения оптимального подхода к терапии, как и детально проанализировать общие тенденции иммунопатогенеза у пациентов по отдельным описаниям клинических случаев.
Заключение
Данный клинический случай предлагает поразмышлять над реклассификацией СПД, т.к. редкие наблюдения позволяют опираться лишь на данные литературы, на основании которых СПД не отличим от аннулярной формы пустулезного псориаза клинически, тем не менее есть несколько патоморфологических характеристик, присутствующих при одном и не выявляющихся при другом состоянии. Поэтому СПД нельзя назвать аннулярным пустулезным псориазом, но можно лишь предположить, что это дополнительный вариант пустулезного псориаза. СПД и субкорнеальный тип IgA-пузырчатки неразличимы ни клинически, ни патоморфологически. IgA-пузырчатка представляет собой интраэпидермальный пузырный дерматоз с циркулирующими и прикрепляющимися к тканям IgA-аутоантителами против антигенов десмосом кератиноцитов. При этом, по данным литературы, циркулирующие антитела класса IgA обнаруживаются в 50% случаев и их обнаружение требует специальных методов диагностики, широко не доступных в медицинской практике. Возможные ответы лежат в плоскости исследований в рамках нескольких дисциплин (медицинской генетики, иммунологии, дерматовенерологии, ревматологии и онкогематологии), но крайне важны, т.к. это обосновало бы подходы к диагностике и терапии таких пациентов.
Согласие пациента. Отсутствует.
