Введение
Ишемическая болезнь сердца (ИБС) является ведущей причиной смерти и инвалидизации населения во всем мире [1]. В связи с увеличением продолжительности жизни и распространенности факторов риска (артериальная гипертензия, курение, нарушение липидного, углеводного и пуринового обменов, ожирение и др.) число пациентов с ИБС неуклонно растет и составляет, согласно обновленным результатам исследования «Global Burden of Disease» [1], 126 млн человек. При этом мужчины болеют чаще, чем женщины, и заболеваемость ИБС увеличивается в соответствии с возрастом [1]. В Российской Федерации, по данным Росстата, заболеваемость ИБС за 2018 г. составила 710,2 на 100 тыс. человек, число летальных исходов от ИБС – 308,7 на 100 тыс. человек [2].
Высокий уровень смертности от ИБС объясняется прежде всего наличием коморбидных состояний, способных взаимно отягощать течение друг друга, поскольку больные ИБС редко когда имеют одно заболевание: чаще наблюдается их сочетание [3]. Кроме того, коморбидность является одним из факторов низкой эффективности лечения, т.к. алгоритмы медикаментозной терапии сочетанных нозологических форм еще до конца не определены [4].
Самой распространенной сопутствующей патологией у пациентов с ИБС считается хроническая сердечная недостаточность (ХСН). Так, по данным международного регистра CLARIFY («prospeCtive observational LongitudinAl RegIstry oF patients with stable coronary arterY disease») [5], у российских пациентов (n=2249) с ИБС частота встречаемости ХСН составила 77,5%.
С другой стороны, согласно популяционному исследованию ЭПОХА-ХСН (n=11453) [6], ИБС среди больных ХСН встречалась в 63,3% случаев, при этом частота ИБС увеличивалась в соответствии с функциональным классом (ФК) ХСН: при I–II ФК 57,5%, при III-IV ФК – 77,9%.
Сочетание ИБС и ХСН значимо ухудшает прогноз и качество жизни пациентов, а также увеличивает риск летального исхода [7–9]. В связи с этим медикаментозная терапия в данной клинической ситуации должна быть не только оптимальной с точки зрения безопасности, но и эффективной [8]. Однако существующие схемы лечения ИБС не привели к значимому снижению частоты приступов стенокардии и госпитализаций, к существенным изменениям в структуре смертности от ИБС, в т.ч. при наличии ХСН [7]. Кроме того, при неудовлетворительном контроле приступов стенокардии существуют некоторые трудности в подборе оптимальной схемы лечения, поскольку добавление основных антиангинальных препаратов, действующих через модификацию гемодинамических изменений (β-адреноблокаторы, блокаторы кальциевых каналов, нитраты), значимо увеличивает риск развития побочных эффектов [8, 9]. Следовательно, в таких ситуациях необходимо использовать другие, альтернативные, методы лечения. В частности, расширение знаний о роли метаболических нарушений и энергодефицита, происходящих при ишемии миокарда, привело к тому, что метаболическая терапия, не оказывающая значимого гемодинамического действия, может рассматриваться в качестве эффективного метода лечения пациентов как c ИБС, так и с сочетанием ИБС и ХСН [10–13].
Стоит отметить, что в настоящее время увеличивается число доказательств, свидетельствующих, что метаболическая недостаточность и митохондриальная дисфункция являются одними из основополагающих механизмов развития и прогрессирования ИБС и ХСН [14–16]. Однако за счет чего происходит активации этих путей, до сих пор полностью не изучено. Например, имеются сведения, согласно которым такой метаболит, как триметиламиноксид (ТМАО), образующийся в печени из триметиламина (эндогенный метаболит микрофлоры кишечника) при потреблении красного мяса и морепродуктов, ассоциирован с ИБС, ХСН и маркерами сердечного гемодинамического стресса [17].
Таким образом, особый интерес в лечении пациентов с ИБС и ХСН представляют лекарственные средства, обладающие гемодинамически нейтральным действием и направленные на повышение энергоэффективности миокарда. Одним из наиболее изученных метаболических препаратов является триметазидин [18, 19].
В связи с этим целью настоящего обзора стало изучение по литературным данным возможности применения триметазидина в лечении пациентов с сочетанием ИБС и ХСН.
Патофизиологические механизмы ХСН при ИБС
ХСН при ИБС возникает в результате гипоксии кардиомиоцитов, вызванной нарушением коронарного кровотока вследствие атеросклеротического поражения сосудов или спазма коронарных артерий [20]. Недостаточное поступление кислорода для удовлетворения метаболических потребностей миокарда приводит к дисфункции желудочков и развитию ХСН [20]. Далее, вследствии снижения сократительной функции миокарда, развития систолических и диастолических нарушений сердечной деятельности различной степени выраженности ткань миокарда начинает подвергаться действию таких стрессовых факторов, как перегрузка объемом или давлением [21]. В данных условиях запускается серия компенсаторных изменений как в кардиомиоцитах, так и в других клетках, окружающих их (фибробласты, эндотелиоциты, гладкомышечные клетки сосудов, иммунокомпетентные клетки) [21]. Большинство взрослых кардиомиоцитов обладают ограниченной способностью к пролиферации и являются терминально дифферинцированными клетками, в то время как клетки соединительной ткани сохраняют способность к пролиферации [22, 23]. Следовательно, адаптация к внешним изменениям происходит у кардиомиоцитов через метаболическое ремоделирование и гипертрофию миокарда, а у фибробластов через пролиферацию в ответ на стресс, приводящую к структурным изменениям в сердце [23].
Под метаболическим ремоделированием миокарда прежде всего понимается смена метаболизма субстратов, используемых клетками миокарда для получения энергии. Физиологически производство аэробного сердечного АТФ происходит за счет митохондриального окисления углеводов и жирных кислот [24]. Для обеспечения высоких энергетических потребностей дополнительные молекулы АТФ могут быть получены через альтернативные субстраты, такие как кетоновые тела [25]. При нормальном функционировании около 70% от всего производства АТФ обеспечивается метаболизмом жирных кислот [25]. У пациентов с ИБС и ХСН миокард не способен производить достаточное количество АТФ для удовлетворения метаболических потребностей, что приводит к энергетическому голоданию [25]. Механизм энергодефицита при ИБС и ХСН довольно сложен и, по-видимому, осуществляется не только за счет недостаточной доступности субстрата, но и за счет нарушения преобразования и утилизации этих субстратов миокардом [20, 26]. При этом снижение производства АТФ усугубляется увеличением потребности миокарда в энергии, обусловленной чрезмерной активацией симпатической нервной системы, которая, как известно, возникает у пациентов с ИБС и ХСН [26].
В начале развития ХСН при ИБС происходит подавление утилизации жирных кислот клетками миокарда с относительным сохранением или увеличением потребления глюкозы [25]. Данный процесс обусловлен снижением трансляционной активности ядерных рецепторов, активируемых пролифератором пероксисом (рeroxisome proliferator-activated receptor, PPAR), и экспрессии ацил-КоА-дегидрогеназы, участвующей в модуляции метаболизма и β-окислении жирных кислот [16, 26]. По мере прогрессирования ХСН при ИБС этот первоначальный ответ приводит к повышению уровня свободных жирных кислот в сыворотке крови, а затем к их накоплению в миокарде в виде триглицеридов [26]. Далее избыток свободных жирных кислот и внутримиокардиальных триглицеридов может значимо ухудшить течение установленных ИБС и ХСН за счет прямой липотоксичности и индуцирования развития инсулинорезистентности [25, 26]. Происходящая параллельно с этим процессом утилизация глюкозы способствует накоплению молочной кислоты и протонов в кардиомиоцитах (вследствии гипоксии при ИБС и ХСН анаэробный гликолиз глюкозы превалирует над ее аэробным окислением), внутриклеточный обмен которых приводит к перегрузке кальцием и развитию митохондриальной дисфункции [27, 28]. Следовательно, на этом этапе ХСН при ИБС большая часть глюкозы, полученная кардиомиоцитами, используется неэффективно, что значимо усугубляет имеющийся энергодефицит. Далее при развитии «сердечной инсулинорезистентности», сопровождающейся снижением инсулинзависимого захвата глюкозы и нарушением метаболизма глюкозы, миокард становится более зависимым от жирных кислот, поэтому метаболический путь утилизации этого субстрата вновь подлежит активации [25]. По сравнению с утилизацией глюкозы окисление свободных жирных кислот требует большей потребности в кислороде и имеет более низкую энергетическую эффективность, что провоцирует прогрессирование ИБС и ХСН [26].
Следующим патофизиологическим механизмом ХСН при ИБС является гипертрофия миокарда. Гипертрофия миокарда – наиболее важное и очевидное изменение структуры сердца при перегрузке объемом или давлением, характеризующееся усилением синтеза белка и увеличением площади поперечного сечения кардиомиоцитов [23]. Повышение давления в полостях сердца активирует работу нейроэндокринной системы, приводящую к усилению выделения в кровь вазоактивных веществ (вазопрессин, эндотелин-1, ангиотензин II), которые, в свою очередь связываясь со своими рецепторами на поверхности кардиомиоцитов, запускают каскад внутриклеточных сигнальных путей (митоген-активированная протеинкиназа, кальмодулинкиназа-ядерный фактор акивированных Т-клеток, N-концевая киназа c-Jun) и способствуют повышению экспрессии генов, ассоциированных с гипертрофией миокарда [29]. При этом стоит отметить, что гипертрофия кардиомиоцитов является адаптивным изменением, способствующим поддержанию систолической функции сердца в период компенсации ХСН при ИБС. В случае, если воздействие фактора (усугубление ишемии, нагрузка давлением или объемом) сохраняется и продолжается длительное время, кардиомиоциты начинают гибнуть в результате некроза, апоптоза и зависимой от гибели клеток аутофагии [30]. Прямое уменьшение числа кардиомиоцитов усугубляет гипертрофию оставшихся клеток миокарда [30]. Снижение числа кардиомиоцитов до определенного предела, когда эти гипертрофированные клетки уже не способны компенсировать сердечную функцию в полном объеме, запускает развитие ХСН. Следовательно, гибель кардиомиоцитов – важный поворотный момент от компенсаторной гипертрофии при ИБС до ХСН.
Другим значимым патофизиологическим механизмом ХСН при ИБС является интерстициальный фиброз миокарда, возникающий вследствие пролиферации фибробластов и диффузного накопления коллагена I и III типов в интерстиции миокарда [31]. Перекрестное связывание коллагена напрямую ухудшает диастолическую функцию миокарда, а его молекулярная перестройка косвенно способствует снижению его сократительной функции за счет снижения проведения сигналов к кардиомиоцитам [32]. За счет увеличения диффузионного расстояния интерстициальный фиброз миокарда значимо ухудшает и так существующую гипоксию кардиомиоцитов и провоцирует энергодефицит [33]. Кроме того, имеются сведения, согласно которым интерстициальный фиброз миокарда способствует развитию желудочковых аритмий при ХСН, т.к. стало известно, что миофибробласты во время межклеточного воздействия способны напрямую действовать на кардиомиоциты и косвенно через паракринные факторы, на синоатриальный узел, влияя тем самым на ритм и проводящую систему сердца, увеличивая риск развития желудочковых нарушений ритма [34].
Так, препараты с многофакторным спектром действия, способные «переключать» метаболизм с жирных кислот на глюкозу, снижать накопление коллагена в интерстиции миокарда, увеличивая потребление кислорода кардиомиоцитами, стимулировать утилизацию глюкозы кардиомиоцитами за счет аэробного окисления, предотвращать апоптоз клеток миокарда, регулировать процессы аутофагии, патогенетически обоснованы для применения при ИБС и ХСН.
Механизмы действия триметазидина при ИБС и ХСН
Триметазидин – это метаболический модулятор, который широко используется при лечении стабильной стенокардии [35, 36] и имеет относительно значительное количество доказательств для его применения при сочетании ИБС и ХСН [37]. Известно, что триметазидин обладает противоишемическими свойствами, не влияя при этом на гемодинамику и потребление миокардом кислорода и не оказывая каких-либо отрицательных инотропных и сосудорасширяющих эффектов [25].
Механизмы действия триметазидина условно можно разделить в соответствии с патофизиологическими изменениями, возникающими при ХСН на фоне ИБС: влияние на энергетический обмен, гибель кардиомио-цитов, развитие интерстициального фиброза миокарда и его воспаление.
Влияние триметазидина на энергетический обмен достигается за счет снижения скорости аэробного окисления жирных кислот путем ингибирования ключевого фермента β-окисления – митохондриального 3-кетоацил-КоА, и косвенной стимуляции утилизации глюкозы посредством увеличения активности пируватдегидрогеназы [38]. Также, учитывая, что при ХСН на фоне ИБС метаболизм глюкозы смещается в сторону гликолиза, необходимо отметить еще один позитивный механизм триметазидина, а именно влияние на молочную кислоту, являющуюся продуктом гликолиза. Данный аспект представляет интерес, поскольку в ряде работ продемонстрировано, что у пациентов с ХСН уровень молочной кислоты не только значимо выше, чем у здоровых лиц [39], но и тесно связан с тяжестью ХСН и прогнозом (высокие уровни молочной кислоты ассоциированы с худшем прогнозом) [40]. По данным Y.H. Fang et al. [41], триметазидин в дозе 70 мг в течении 8 недель увеличивал сердечный выброс и переносимость физической нагрузки у крыс с гипертрофией правого желудочка, а также снижал уровень молочной кислоты и увеличивал аэробное окисление глюкозы. В другом экспериментальном исследовании (модель ишемии сердца у крыс) показано, что триметазидин не только способствует постишемической репарации, но и снижает скорость гликолиза, повышает интенсивность аэробного окисления глюкозы, что важно при сочетании ИБС и ХСН [42]. Также триметазидин поддерживает внутриклеточные концентрации фосфокреатина и АТФ и улучшает их соотношение при ИБС и ХСН, увеличивая при этом ресинтез фосфокреатина и восстановление митохондриального окислительного фосфорилирования [43].
Гибель кардиомиоцитов, как было описано ранее, является ключевым поворотным фактором от компенсации до декомпенсации ХСН при ИБС. В экспериментальной работе (модель ХСН) триметазидин значимо предотвращал гибель кардиомиоцитов, реализуя кардиопротективное действие [44]. Механизмы данного эффекта объясняются тем, что триметазидин способен активировать микро-РНК (miR-21), запускающую сигнальный путь блокировки апоптоза клеток миокарда, и снижать продукцию активных форм кислорода, оказывающих прямое цитотоксическое действие [45, 46]. Кроме того, триметазидин может модулировать аутофагию кардиомиоцитов, индуцируя умеренную и ингибируя чрезмерную аутофагию, тем самым способствуя функциональному восстановлению клеток миокарда [47, 48].
Ключевым фактором при интерстициальном фиброзе миокарда, вызывающем секрецию внеклеточного матрикса и пролиферацию фибробластов, является фактор роста соединительной ткани (connective tissue growth factor, CTGF). Интересно, что триметазидин эффективно подавляет как накопление коллагена I и III типов в миокарде, так и экспрессию CTGF [49]. Улучшая структуру миокарда, триметазидин способствует снижению выраженности нарушений сократительной способности и повышению эластичности миокарда, увеличению эффективности использования кислорода кардиомиоцитами и улучшению симптомов ИБС и ХСН. При этом важно подметить, что триметазидин при фиброзе миокарда оказывает влияние не только на кардиомиоциты, но и на клетки, окружающие их.
Еще одним важным механизмом действия триметазидина является влияние на воспалительные процессы, которые развиваются при ИБС и ХСН (индукция фактора некроза опухоли-α, интерлейкинов-6, -18 и т.д.). По данным X. Zhou et al. [50], триметазидин значимо снижал уровни таких воспалительных маркеров в сыворотке крови, как интерлейкин-1β, интерлейкин-6 и фактор некроза опухоли-α в модели ремоделирования левого желудочка у крыс [50].
Клинические исследования триметазидина при сочетании ИБС и ХСН
Многие рандомизированные контролируемые исследования (РКИ) и мета-анализы подтвердили защитное действие триметазидина при сочетании ИБС и ХСН, заключающееся в улучшении клинических проявлений и сердечной функции (данные суммированы в таблице). Что касается механизмов, подтвержденных не в фундаментальных исследованиях (описаны ранее), а в клинических, то триметазидин снижает экспрессию предсердного натрийуретического пептида [51], повышает уровень высокоэнергетического фосфата в левом желудочке [52], а за счет уменьшения вариабельности сердечного ритма и сокращения корригированного интервала QT снижает риск развития нарушений ритма при ИБС и ХСН [53].
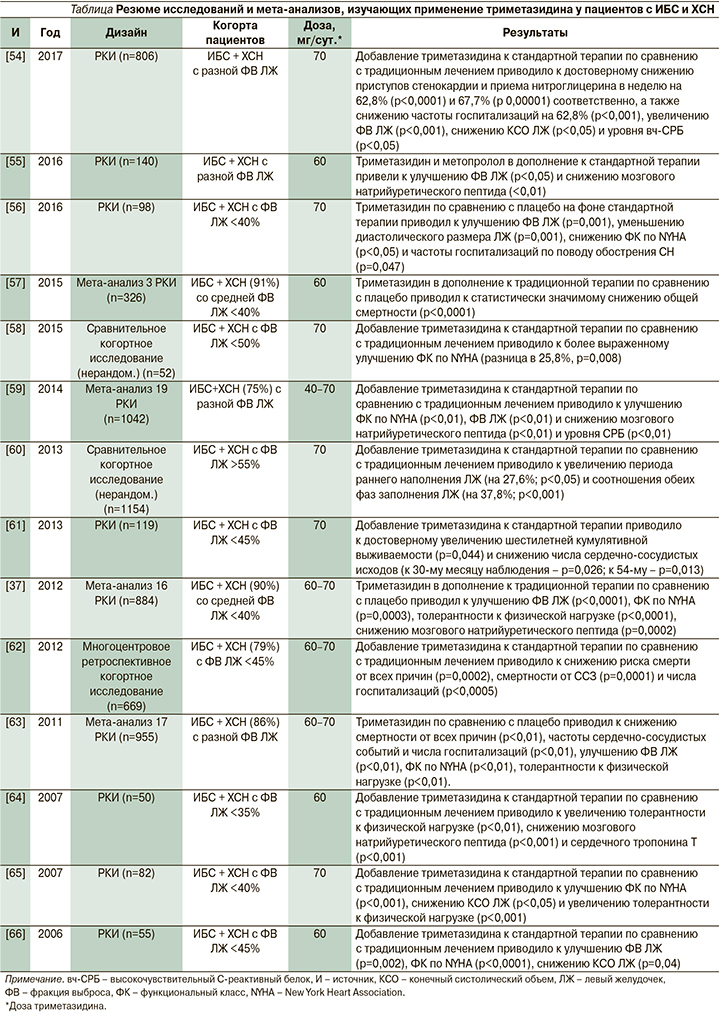
Важным аспектом применения триметазидина при сочетании ИБС и ХСН является не только выбор оптимальной дозы данного препарата (как видно из таблицы в большинстве исследований применялся триметазидин в дозе 70 мг/сут.), но и продолжительность его приема. В клинических исследованиях время лечения пациентов с ИБС и ХСН триметазидином сильно варьируется и составляет от 1 до 48 месяцев. Однако наибольший позитивный эффект, как правило, наблюдается при длительности терапии 3–6 месяцев и более. Так, в исследовании, проведенном А. Momen et al. [56], триметазидин в дозе 70 мг в сутки в течение 6 месяцев улучшал не только функцию сердца, но и качество жизни пациентов, повышая толерантность к физической нагрузке и снижая частоту госпитализаций по поводу обострения ХСН. Эти данные показывают, что чем больше продолжительность лечения триметазидином, тем значительнее улучшение сердечной функции и прогноз. Кроме того, стоит отметить, что помимо улучшения фракции выброса ЛЖ триметазидин может также улучшать эндотелий-зависимую релаксацию сосудов, что важно в лечении пациентов с ИБС и ХСН [67].
На основании клинической эффективности триметазидина у пациентов с ИБС и ХСН данный препарат поддерживается текущими клиническими рекомендациями по ИБС и ХСН.
Позиция триметазидина в клинических рекомендациях по ИБС и ХСН
Несмотря на отсутствие ХСН как зарегистрированного показания к применению триметазидина, в российских клинических рекомендациях «Стабильная ишемическая болезнь сердца» (2020) [11] и рекомендациях Европейского общества кардиологов по диагностике и лечению хронического коронарного синдрома (2019) [12], применение триметазидина в качестве препарата второй линии возможно не только при наличии недостаточной эффективности препаратов первой линии у пациентов со стабильной стенокардией, но и при сочетании стенокардии с ХСН.
В то же время в российских клинических рекомендациях «Хроническая сердечная недостаточность» (2020) [9] отмечается, что применение триметазидина пациентами с ишемической ХСН безопасно и эффективно, поэтому при сохранении приступов стенокардии для усиления антиангинального эффекта следует добавить триметазидин к традиционной терапии.
Также, согласно обновленным Клиническим рекомендациям по диагностике и лечению острой и хронической СН Европейского общества кардиологов (2021) [12], триметазидин может быть использован в оношении пациентов с ишемической ХСН и стенокардией, несмотря на применение β-адреноблокаторов и/или ивабрадина, поскольку он значимо улучшает функцию ЛЖ и переносимость физических нагрузок пациентами с ХСН и хроническим коронарным синдромом.
Заключение
Применение триметазидина при ИБС и ХСН является не только патогенетически обоснованным, но и клинически эффективным с точки зрения улучшения функции сердечно-сосудистой системы, качества жизни пациентов и прогноза. А поскольку большинство исследований, в т.ч. длительных, ориентировано в данной клинической ситуации преимущественно на суточную дозу триметазидина в 70 мг при лечении пациентов с ИБС и ХСН этой дозе следует отдавать предпочтение. На российском рынке доступен и широко используется такой препарат триметазидина пролонгированного действия в дозе 70 мг, как Депренорм® ОД (ЗАО «Канонфарма продакшн», Россия), безопасность и эффективность которого подтверждена в клинических исследованиях. Также Депренорм® ОД (ЗАО «Канонфарма продакшн», Россия) имеет доказанную биоэквивалентность оригинальному препарату триметазидина.
Терапевтическая биоэквивалентность Депренорм® (ЗАО «Канонфарма продакшн», Россия) оригинальному препарату триметазидина изучалась в многоцентровом открытом рандомизированном исследовании КАРДИОКАНОН [68], включившем 120 пациентов с ИБС и стабильной стенокардией напряжения II–III ФК (период наблюдения – 12 недель).
В результате показано, что между препаратом Депренорм® (ЗАО «Канонфарма продакшн», Россия) и оригинальным препаратом триметазидина отсутствовали различия в антиангинальных эффектах, заключающихся в сопоставимом снижении частоты приступов стенокардии в неделю (р=0,10) и приема таблеток нитроглицерина в неделю (р=0,72). Кроме того, по завершении исследования у пациентов наблюдалось улучшение ФК стенокардии, что свидетельствует о повышении качества жизни пациентов с ИБС: I ФК – частота увеличилась с 0 до 61,5%, II ФК – частота снизилась с 97,5 до 38,5%, III ФК – частота снизилась с 2,5 до 0% [68].
Таким образом, препарат Депренорм® ОД (ЗАО «Канонфарма продакшн», Россия) имеет доказанную терапевтическую эквивалентность и биоэквивалентность оригинальному препарату триметазидина. Вместе с тем лекарственная форма Депренорм® ОД (ЗАО «Канонфарма продакшн», Россия) в виде таблеток с пролонгированным высвобождением позволяет использовать его один раз в сутки, что значимо повышает уровень комплаентности пациентов к лечению.
Конфликт интересов. Статья подготовлена при поддержке компании ЗАО «Канонфарма продакшн».
