Амилоидоз сердца (АС) – одно из самых тяжелых проявлений системного амилоидоза. При первичном амилоидозе выраженное поражение сердца ведет к снижению средней выживаемости до 6 месяцев, тогда как при преимущественном поражении периферической нервной системы этот показатель
составляет около 4 лет. При других формах системного амилоидоза поражение сердца также существенно ухудшает прогноз.
Классификация и эпидемиология
При АС происходит инфильтрация миокарда нерастворимым гликопротеидом – амилоидом, образующимся из различных сывороточных или локально продуцируемых белковпредшественников. В их число входят константные белки, не зависящие от типа амилоида, например сывороточный амилоидный Р-белок (SAP), однако около 80 % массы амилоида составляют белки, отличающиеся при
разных типах амилоида. Современная классификация амилоидоза основана на различиях амилоидогенных белков-предшественников. Таких белков в настоящее время известно около 30;
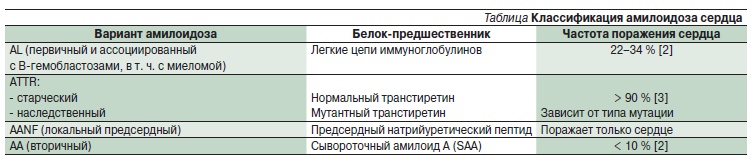
соответственно, выделяют около 30 форм амилоидоза. Каждую форму обозначают аббревиатурой, включающей обозначение белка-предшественника: AL (L – легкие цепи иммуноглобулинов), ATTR (TTR – транстиретин), СAA (сывороточный амилоид А) и др. (см. таблицу). Согласно этой классификации, первичный и ассоциированный с миеломой амилоидоз следует обозначать аббревиатурой AL, т. к. амилоид при этих формах образован легкими цепями иммуноглобулинов. При системных формах амилоидоза (большинство случаев AL, а также ATTR, AA и др.) амилоидогенный белок-предшественник циркулирует в крови и может откладываться в различных тканях, тропность к которым отличается у разных белков. Кроме того, возможно развитие локального амилоидоза, например предсердий,
вследствие местной продукции амилоидогенного варианта предсердного натрийуретического фактора (AANF).
Таблица. Классификация амилоидоза сердца.
Самым частым вариантом системного амилоидоза считают AL-амилоидоз – одну из многочисленных форм плазмоклеточной дискразии. Термин “плазмоклеточная дискразия” объединяет группу состояний, обусловленных патологией плазматических клеток костного мозга, реже – других тканей. При этом в костном мозге отмечается доминирование патологического клона плазматических клеток, синтезирующих аномальные иммуноглобулины или их компоненты: тяжелые или легкие цепи. Кроме AL-амилоидоза к плазмоклеточным дискразиям относят множественную миелому, макроглобулинемию Вальденстрема и др.
При AL-амилоидозе патологический клон плазматических клеток синтезирует легкие цепи иммуноглобулинов, осаждение которых в тканях сопровождается образованием нерастворимых
фибрилл амилоида. AL-амилоидоз – наиболее тяжелая, генерализованная форма заболевания, поражающая многие ткани. Сердце поражается у 90 % больных, у половины из которых развивается диастолическая сердечная недостаточность (СН) [2]. Смерть обычно наступает от СН или аритмий [4].
Изолированный кардиопатический вариант AL-амилоидоза диагностируют редко – менее чем у 5 % больных [5]. Изолированное поражение сердца характерно для AANF-амилоидоза, при котором отмечают инфильтрацию предсердий амилоидом, образующимся из предсердного натрийуретического пептида. Эту форму амилоидоза обнаруживают более чем у 95 % людей старше 80 лет [6]. AANF-амилоидоз редко приводит к СН [2], но может стать причиной развития наджелудочковых аритмий [7–9].
Поражение сердца характерно для приобретенного и различных вариантов наследственного ATTR-
амилоидоза. Эта форма обусловлена отложением белка-переносчика гормонов щитовидной железы и ретинола – транстиретина. Он секретируется печенью в тетрамерной форме, состоящей из четырех молекул транстиретина. С возрастом снижение активности ферментных систем гепатоцита приводит к повышению секреции амилоидогенных мономерных форм транстиретина и вследствие этого – раз-
витию амилоидоза. Приобретенный ATTR-амилоидоз обычно называют системным старческим амилоидозом (поражает до 25–36 % людей старше 80 лет). Амилоид находят во многих органах, однако наиболее значимые депозиты обнаруживают в сердце. Обычно заболевание имеет доброкачественное малосимптомное течение, проявляется блокадой передней ветви левой ножки пучка Гиса и небольшим утолщением стенок левого желудочка (ЛЖ). При значительном отложении амилоида могут развиваться кардиомегалия и медленнопрогрессирующая СН [3].
При наличии мутаций в молекуле транстиретина сборка тетрамеров в гепатоците нарушена исходно, что приводит к развитию наследственного ATTR-амилоидоза в более раннем возрасте. В настоящий момент известно около 100 точечных мутаций и делеций гена транстиретина, 87 из которых амилоидогенны [10]. Для заболевания характерно аутосомно-доминантное наследование, однако пенетрантность мутантного гена варьируется в широких пределах. Выделяют невропатическую, кардиопатическую и офтальмолептоменингеальную формы заболевания. Чаще всего развивается невропатическая форма, которая характеризуется прогрессирующим поражением периферической и автономной нервной системы. Преимущественное поражение того или иного органа во многом связано с характером мутации. Так, при замене изолейцина в позиции 122 на валин развивается
старческое амилоидное поражение сердца без неврологических проявлений; эту мутацию обнаруживают преимущественно у афроамериканцев [11]. К мутациям, вызывающим серьезное поражение сердца, относят замену метионина на валин в положении 30 (португальский вариант семейного амилоидоза), замену серина на изолейцин в положении 84 и замену аланина на треонин в положении 60. Амилоидная кардиопатия при наследственном ATTR-амилоидозе протекает менее тяжело, чем при AL-амилоидозе, но все же приводит к выраженной СН. При одном из наиболее распространенных вариантов амилоидоза – вторичном АА-амилоидозе, клинически значимое поражение сердца обнаруживают редко – менее чем в 10 % случаев [13]. Хотя АА-амилоидоз имеет
системный характер, а при морфологическом исследовании депозиты амилоида обнаруживают в различных тканях, при этой форме заболевания клиническое значение имеет в основном поражение почек, в меньшей степени – печени, селезенки и желудочнокишечного тракта.
Морфология и патогенез
Отложение амилоида приводит к значительному концентрическомуутолщению миокарда, причем для АС не характерна дилатация полостей желудочков. Амилоидная инфильтрация миокарда сопровождается ухудшением его механических свойств и приводит к значительному снижению
сократимости в сочетании с тяжелыми рестриктивными нарушениями диастолической функции. Современные методы исследования показали, что утолщение стенок миокарда при АС не всегда коррелирует с нарушением систолической и диастолической функций. Условно можно выделить
3 подтипа АС – у больных AL, наследственным ATTR и приобретенным сенильным ATTR-амилоидозом [16].
Во всех случаях амилоид обнаруживают в интерстиции миокарда в виде диффузных или узловых депозитов. Наиболее часто выявляют диффузное утолщение межжелудочковой перегородки (> 80 % больных) и задней стенки ЛЖ. Изолированное утолщение стенки правого желудочка (без утолщения стенки ЛЖ) встречается редко – в 6 % случаев, а утолщение стенок обоих желудочков – в 40–80 % случаев [3, 18]. Утолщение межпредсердной перегородки отмечается у 40 % больных. Изменение формы ЛЖ происходит по типу концентрической гипертрофии, дилатация желудочков не развивается [19]. Сочетание утолщения стенок миокарда более чем у половины больных с низкой амплитудой
желудочкового комплекса на электрокардиограмме (ЭКГ) в грудных отведениях (менее 10 мм) и отведениях от конечностей (менее 5 мм) позволяет подтвердить псевдогипертрофический характер изменений миокарда. По данным C. Rapezzi и соавт., толщина стенок миокарда у больных ATTR-амилоидозом (особенно старческим) была достоверно больше, чем у пациентов с AL-амилоидозом [16, 20]. В то же время у больных AL-амилоидозом отмечено более значительное снижение амплитуды желудочкового комплекса и, соответственно, более низкое отношение амплитуды QRS к
толщине стенки ЛЖ. Это может указывать на связь снижения амплитуды QRS у больных AL-амилоидозом в бóльшей степени с повреждением кардиомиоцитов, чем с интерстициальной депозицией амилоида. Известно, что в патогенезе повреждения органов у больных AL-амилоидозом большое значение имеет прямое токсическое действие свободных легких цепей иммуноглобулинов на клетки [16, 18]. У больных с разными типами АС обнаруживают значительную вакуолизацию кардиомиоцитов. Исследования последних лет показали [24], что именно вакуолизацией кардиомиоцитов, а не депозитами амилоида обусловлена повышенная эхогенность зернистого
типа, характерная для АС и впервые описанная А. Siqueira-Filho и соавт. [19]. Таким образом, это ультразвуковое свойство амилоидного сердца не является специфичным и не может быть использовано в качестве диагностического критерия.
Отражением повреждения кардиомиоцитов у больных АС может быть повышение концентрации тропонинов и натрийуретических пептидов. Было показано, что экспрессия предсердного и мозгового нитрийуретических пептидов повышена в кардиомиоцитах желудочков у пациентов с АС, особенно в участках, прилегающих к депозитам амилоида [25]. Хотя указанные субстанции не являются специфическими маркерами амилоидного поражения сердца, их считают высокочувствительными показателями и, следовательно, важным фактором прогноза при системном амилоидозе [26].
Амилоид может откладываться в области клапанов сердца, часто вызывая их утолщение, заметное на эхокардиограмме – Эхо-КГ [18, 27]. Описано утолщение папиллярных мышц [28], однако функция клапанов чаще сохранена.
Амилоид способен поражать проводящую систему сердца, описаны случаи проникновения амилоидных масс непосредственно в синоатриальный узел. Нарушения проводимости чаще всего представлены неполной блокадой передней ветви левой ножки пучка Гиса (20 %), полной блокадой правой
(4–19 %) или левой ножки (2–7 %), атриовентрикулярной блокадой I степени (18–33 %) [16, 18]. Нарушения проводимости, как правило, встречаются при старческом АС, что, возможно, связано с более длительным течением данного варианта заболевания [3]. Отложение амилоида в области адренергических синапсов нарушает нейрогуморальную регуляцию работы сердца [29], что может быть одним из факторов риска тахиаритмий. У 5–27 % больных выявляют фибрилляцию предсердий, более редкими нарушениями ритма являются желудочковая тахикардия, узловой ритм. Какие-либо
изменения на ЭКГ встречаются почти у всех (> 90 %) больных АС.
Отличительным свойством AL-амилоидоза является его отложение в стенках коронарных артерий [15], что может вызывать ишемию миокарда [30]. Некоторые больные жалуются на стенокардию [31], при этом изменения на ангиограмме могут отсутствовать [32]. Может развиваться инфаркт миокарда. Патологические Q-зубцы и изменения реполяризации без клинических признаков инфаркта миокарда
или указания на него в анамнезе обнаруживают более чем у половины больных, что позволяет интерпретировать эти изменения как псевдоинфарктные. По-видимому, имитация инфарктных изменений связана с узловым отложением электрически неактивного амилоида, что создает эффект рубца. На поздних стадиях заболевания почти у половины больных выявляют перикардиальный выпот [18].
Количественная оценка совокупной массы амилоидных депозитов в сердце возможна с помощью SAP-
сцинтиграфии. SAP – это нормальный гликопротеин плазмы, уровень которого резко возрастает в амилоидных депозитах любого типа в результате обратимого кальций-зависимого связывания с амилоидной фибриллой. После внутривенного введения радиофармпрепарат с меченым SAP рас-
пределяется между циркулирующим и связанным с амилоидом пулами SAP пропорционально их объему. Таким образом, можно получить изображения для качественной и количественной оценки амилоидных депозитов. Метод особенно полезен при мониторировании течения амилоидоза, в т. ч. с целью оценки эффективности лечения.
Нарушения функции сердца
Нарушения внутрисердечной гемодинамики при АС обусловлены главным образом диастолической дисфункцией, снижение сократительной способности миокарда обычно незначительное. В исследовании, включившем 223 больных АС, средняя фракция выброса ЛЖ составила 52,5 ± 13,1 %
у больных AL-амилоидозом и 58 ± 13 % у пациентов с наследственным ATTR-амилоидозом. Она была достоверно ниже только среди больных старческим ATTR-амилоидозом 44,2 ± 15,4 % [16]. Снижение фракции выброса менее 40 % было отмечено у 40 % больных старческим ATTR-амилоидозом и только у 22 % пациентов с AL-амилоидозом и 8 % больных наследственным ATTR-амилоидозом. Сходные изменения сократительной функции описывают и другие исследователи [18].
Диастолическая дисфункция разной степени определяется практически для всех пациентов. На ранних этапах развития болезни отложения амилоида нарушают изоволюмическое расслабление миокарда, что приводит к уменьшению скорости раннего диастолического наполнения ЛЖ (E) и увеличению позднего диастолического потока (А). Таким образом, снижение отношения Е/А (1-й тип диастолической дисфункции) является ранним признаком поражения сердца. При прогрессировании АС стенка миокарда становится более жесткой, давление в левом предсердии возрастает, что приводит к увеличению скорости раннего диастолического наполнения и, таким образом, к псевдонормализации отношения Е/А (2-й тип диастолической дисфункции) [2]. При дальнейшем
увеличении жесткости сердечной стенки и росте конечного диастолического давления происходит значительное снижение позднего диастолического потока, что позволяет выявить снижение растяжимости миокарда (3-й рестриктивный тип диастолической дисфункции). Рестриктивные изменения часто сопровождаются расширением обоих предсердий [3, 18].
Хотя рестриктивные нарушения диастолической функции считают наиболее типичными для АС, данные исследований показывают возможность развития и других нарушений внутрисердечной гемодинамики [34–36].
Клиническая картина и прогноз
Амилоидное поражение сердца на ранних стадиях может протекать бессимптомно, проявляясь лишь утолщением стенки ЛЖ при Эхо-КГ [37]. В дальнейшем развиваются клинические проявления СН с застоем крови преимущественно по большому кругу кровообращения [18]. Прогрессирующая одышка, снижение толерантности к физическим нагрузкам отмечаются более чем у половины пациентов и сопровождаются признаками легочной гипертензии [2, 38]. При AL- и старческом ATTR-амилоидозе
СН встречается чаще (III–IV функциональные классы по NYHA более чем у половины пациентов), чем при наследственном ATTR-амилоидозе [3, 16]. Системность поражения характерна для AL-амилоидоза. У большинства развиваются поражения почек (нефротический синдром, почечная недостаточность), печени (гигантская гепатомегалия, внутрипеченочный холестаз), селезенки (спленомегалия), мышц (макроглоссия, псевдогипертрофия мышц), кожи (периорбитальная пурпура, амилоидные бляшки),
периферической и автономной нервной системы. Изредка развиваются тромбоэмболии коронарных артерий амилоидными массами, приводящие к смерти [39]. Описаны также тромбозы различных сосудов [18].
Диагноз системного амилоидоза устанавливают при гистологическом исследовании ткани, полученной при биопсии. Чаще исследуют стенку прямой кишки, желудка, полости рта, подкожную жировую клетчатку, ткань почки, печени. Определение варианта амилоидоза проводят иммуногистохимическим методом, при наследственном амилоидозе делают анализ ДНК. Если амилоидоз ограничен поражени-
ем сердца, как, например, при наследственном ATTR-амилоидозе с мутацией изолейцина в положении 122, то единственным методом диагностики является биопсия миокарда. Однако вбольшинстве других случаев диагноз АС может быть установлен при обнаружении амилоида в биоптате другого органа или ткани и выявлении у больного утолщения стенки ЛЖ более 12 мм по данным Эхо-КГ [40].
Наиболее тяжелый прогноз – при AL-амилоидозе, несколько более благоприятный – при ATTR-амилоидозе. Причем прогноз определяет именно поражение сердца. В исследовании 232
больных AL-амилоидозом с поражением сердца средняя продолжительность их жизни после установления диагноза составила 1 год. Наличие СН снижало выживаемость: 0,75 против 2,34 года у
больных без СН.
Традиционно кардиальными предикторами неблагоприятного прогноза помимо тяжести СН и рестриктивных нарушений диастолической функции ЛЖ считают утолщение и увеличение массы ЛЖ со снижением фракции выброса [18, 41]. В то же время C. Rapezzi и соавт. выявили наиболее значительное утолщение стенки миокарда и увеличение массы миокарда у больных старческим ATTR-
амилоидозом, при котором отмечается относительно медленное прогрессирование АС [16]. Примечательно, что умеренное утолщение стенок миокарда у больных AL-амилоидозом, участвовавших в этом исследовании, сочеталось со значительным снижением вольтажа желудочковых комплексов
ЭКГ. Авторы связали эти изменения на ЭКГ с прямым токсическим эффектом легких цепей иммуноглобулинов на кардиомиоциты. В то же время массивные депозиты амилоида у больных старческим ATTR-амилоидозом, по-видимому, не оказывают столь неблагоприятного действия на клетки миокарда и, следовательно, меньше влияют на выживаемость больных. В другом исследовании 57 больных с AL- и ATTR-амилоидозом низкий вольтаж ЭКГ также сопровождался ухудшением прогноза [42].
В настоящее время важным предиктором неблагоприятного прогноза считают высокие уровни в крови маркеров повреждения кардиомиоцита – тропонинов T и I, N-концевого пробелка мозгового натрийуретического фактора (NT-proBNP) в момент установления диагноза АС. Так, у 261 больного с впервые установленным диагнозом средняя продолжительность жизни при повышении уровня тропонинов I или T составила соответственно 6 и 8 месяцев, а в отсутствие тропонинов I или T в крови – 21 и 22 месяца [43]. В этом исследовании прогностическое значение уровня тропонинов сыворотки было выше, чем значение застойной СН и эхокардиографических параметров. Изучается возможность анализа динамики уровня NT-proBNP после лечения для оценки прогноза АС. При AL-амилоидозе снижение уровня NT-proBNP на 30 % после химиотерапии ассоциировалось с улучшением прогноза [44].
Важным прогностическим фактором являются также наличие и тяжесть внесердечных проявлений системного амилоидоза (ортостатическая гипотония, осложнения нефротического синдрома и моторной диареи, почечная недостаточность и др.).
Таким образом, среди вариантов АС наиболее тяжелым и быстропрогрессирующим течением отличается AL-амилоидоз, при котором развитие СН мало зависит от степени утолщения стенок миокарда и не всегда сочетается с рестриктивными нарушениями диастолической функции. Прогрессирование АС у этих больных в бóльшей степени обусловлено повреждением кардиомиоцита. У больных старческим ATTR-амилоидозом отмечается относительно мягкое течение кардиопатии, несмотря на значительные структурные (выраженную псевдогипертрофию) и функциональные (рестриктивные расстройства гемодинамики) нарушения. Промежуточное положение занимает наследственный ATTR-амилоидоз, при котором симптомы кардиопатии близки к таковым AL-амилоидоза сердца, однако отличаются меньшей злокачественностью.
Лечение
Одной из основных целей лечения больных АС является снижение количества амилоидогенных белковпредшественников в крови и, таким образом, остановка роста депозитов амилоида. Лечение системного амилоидоза зависит от варианта заболевания. Так, при AL-амилоидозе снижение количества белков-предшественников, т. е. легких цепей иммуноглобулинов, достигается элиминацией патогенного клона плазматических клеток при помощи химиотерапии, в т. ч. высокодозных режимов с поддержкой аутологичными стволовыми клетками костного мозга [2]. Наиболее приемлемой схемой лечения в настоящее время считают схему MDa (мелфалан в низких дозах в сочетании с дексаметазоном в умеренно высоких дозах ежемесячными курсами), а также лечение ингибитором протеасом (бортезомиб).
При наследственном ATTR-амилоидозе трансплантация печени, в которой синтезируется практически
весь транстиретин организма, ведет к полному прекращению продукции мутантного амилоидогенного транстиретина и является единственным на данный момент методом лечения. Трансплантация печени была проведена более 500 больным с ATTR-амилоидозом [45]. Пятилетняя выживаемость составила от 60 до 77 %, при этом отмечена положительная динамика в течении невропатии [46, 47]. При значительных депозитах амилоида в сердце у больных ATTR-амилоидозом было проведено несколько успешных комбинированных трансплантаций сердца и печени [49].
Многие стандартные препараты нельзя применять для лечения СН и аритмий при АС. При СН у этой
категории больных по-прежнему чаще всего используют диуретики, в т. ч. в высоких дозах при сопутствующем нефротическом синдроме. Применение ингибиторов ангиотензинпревращающего фермента и блокаторов рецепторов ангиотензина II при АС ограничено [50]. Ортостатическая гипотония и облитерация амилоидом симпатических рецепторов в сердце ограничивают и использование β-адреноблокаторов. При применении антагонистов кальция описаны случаи ухудшения течения СН [51, 52]. Дигоксин вызывал при АС нарушения сердечного ритма и остановку сердца.
Трудности возникают и при лечении аритмий при АС. Бетаадреноблокаторы иногда с осторожностью назначают на ранних стадиях поражения сердца. Однако их применение в ретроспективном исследовании ассоциировалось с ухудшением выживаемости [55]. У больных с ортостатической гипотонией для повышения артериального давления можно применять мидодрин. При фибрилляции предсердий возможно лечение амиодароном, который применяли также при неустойчивой желудочковой тахикардии [56].
Обсуждается возможность трансплантации сердца как метода лечения АС [57, 58]. Однако продолжительность жизни пациентов после трансплантации остается невысокой, что является следствием рецидива болезни в трансплантате. Чаще всего смерть после операции наступала вследствие амилоидоза других органов или инфекционных осложнений [59].
Изучение механизмов амилоидогенеза позволяет разрабатывать новые подходы к лечению заболевания. Сегодня активно изучаются молекулылиганды, стабилизирующие тетрамерную структуру транстиретина и предотвращающие образование амилоидных фибрилл. Их применяют для лечения
ATTR-амилоидоза и профилактически среди больных с мутациями гена трастиретина.

{kind=link}