Введение
Внебольничная пневмония (ВП) остается распространенным и потенциально жизнеугрожающим заболеванием. Так, число летальных исходов от пневмонии и ее осложнений превышает число больных, умерших от туберкулеза, СПИДа, инфекционного эндокардита и менингита вместе взятых [1, 2]. Ежегодно только в пяти европейских странах (Великобритания, Франция, Италия, Германия, Испания) диагностируется около 3 млн случаев заболевания, а в США эта цифра достигает 5–6 млн [3, 4]. В России (согласно расчетным данным) общее число больных ВП в возрасте ≥18 лет превышает 1,5 млн человек в год [5].
Не вызывает сомнений тот факт, что своевременное и эффективное управление ВП, а именно: а) проведение рентгенографии органов грудной клетки в течение первых 4 часов после клинического дебюта/обращения за медицинской помощью; б) ранняя и адекватная антибактериальная терапия (АБТ), охватывающая основные возбудители заболевания; в) целенаправленное выявление пациентов, нуждающихся в респираторной и гемодинамической поддержке и, таким образом, требующих ведения в отделении интенсивной терапии; г) оценка неблагоприятного прогноза с использованием соответствующих прогностических шкал, связывают с улучшением исходов заболевания [6]. Ну а, поскольку успех лечения ВП зависит от неотложного применения антибиотиков, эффективных в отношении вероятных возбудителей, врачи должны учитывать тяжесть течения заболевания и преобладающие региональные особенности антибиотикорезистентности в процессе выбора соответствующей лечебной тактики. При этом важный вопрос в процессе достижения эффективного управления ВП заключается в том, являются ли существующие антибиотики адекватным «инструментом» лечения и существует ли потребность в новых противомикробных препаратах [7, 8]? Ответ на этот вопрос тем более важен, что по свидетельству современных эпидемиологических исследований стартовая АБТ, не обладающая активностью в отногении микробов-возбудителей (т.н. неадекватная эмпирическая АБТ), корреспондирует с большей смертностью среди больных ВП [9, 10]. Причины же неадекватной АБТ состоят в первую очередь в неспособности охватить конкретный возбудитель ВП или предполагают «участие» в патологическом процессе бактериального патогена, устойчивого к предписанному режиму лечения [11–13].
Открытие новых антибиотиков является чрезвычайно сложной задачей [14, 15]. Во всех других областях медицины существует положительная корреляция между накопленными знаниями и открытием новых лекарственных средств. Эта, казалось бы, очевидная связь парадоксальным образом перевернута для антибиотиков. Когда-то эта область переживала «золотую эру» открытий, начатую исследованиями Selman Waksman (Lewis, 2012), показавшего способность почвенных актиномицетов ингибировать зоны роста тестируемых микробов на чашке Петри [16]. Это привело к открытию стрептомицина [17], а затем за относительно короткий срок последовало открытие и других основных классов антибиотиков (см. рисунок). Впрочем, «золотая эра» закончилась довольно резко в начале 1960-х гг., как раз когда начали накапливаться знания о механизмах действия антибиотиков и природе антибиотикорезистентности. Некогда надежная платформа S. Waksman теперь все чаще открывала известные соединения – аминогликозиды, тетрациклины, β-лактамы, хлорамфениколы и макролиды. Примерно в то же время были достигнуты значительные успехи и в открытии синтетических противомикробных препаратов (изониазид, пиразинамид, этамбутол, метронидазол, фторхинолоны), а позже и превращение соединений узкого спектра действия против грамположительных видов в антибиотики широкого спектра действия (пенициллин →ампициллин; эритромицин →азитромицин) и аналоги, активные в отношении резистентных патогенов [19].
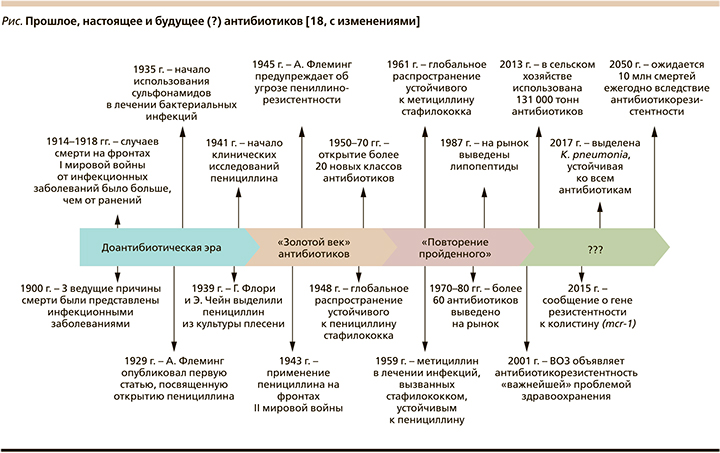
В отличие от других областей медицины уникальность лечения инфекционных заболеваний заключается в необходимости уничтожения живого организма. Естественно, что последствия противодействия соответствующим лечебным вмешательствам не заставляют себя долго ждать и вслед за применением антибиотика неотступно как тень следует лекарственная устойчивость, ограничивающая возможности АБТ [20]. В этом и состоит уникальность антибиотиков, являющихся единственным классом лекарственных средств, демонстрирующих «трансмиссивную потерю эффективности с течением времени» [21].
Примечательно при этом то обстоятельство, что со временем интервал между открытием того или иного антибиотика и выделением культуры устойчивого к нему возбудителя драматически сокращается. Так, например, если с начала активного клинического применения пенициллинов, макролидов, цефалоспоринов до первых сообщений об устойчивых к ним штаммах Streptococcus pneumoniae проходили десятилетия, то время с момента официального одобрения левофлоксацина FDA (Food and Drug Administration) для лечения инфекций дыхательных путей и до выделения устойчивых к нему пневмококков практически совпало [22].
Это обстоятельство во многом сдерживало интерес (прежде всего коммерческий) фармацевтических компаний к разработке новых антибиотиков, и скорость распространения резистентности стала значительно превосходить скорость открытия препаратов, ее преодолевающих.
В связи с этим потребовались беспрецедентные усилия на государственном и межгосударственном уровнях, чтобы «вернуть» интерес фармкомпаний к созданию новых антибиотиков, что, в частности, анонсировалось в 2010 г. инициативой «10×20» (создание 10 новых антибиотиков к 2020 г.) [23].
Усилия по возвращению «утраченного интереса» не оказались бесплодными, и в течение 10 последующих лет мы стали обладателями ряда новых и весьма перспективных антибиотиков, некоторые из которых были предложены и для лечения ВП (табл. 1).

Цефалоспорины: цефтаролин
Цефалоспорины известны своим широким спектром действия, доказанной эффективностью и благоприятным профилем безопасности, что делает их одними из наиболее часто назначаемых противомикробных препаратов. Цефтаролин (активный метаболит цефтаролина фосамила) – новый цефалоспорин широкого спектра действия, отнесенный к V поколению этого класса антибиотиков, отличительными особенностями которого являются активность в отношении метициллиноустойчивых штаммов Staphylococcus aureus (MRSA) и Streptococcus pneumoniae, устойчивых к пенициллину и другим цефалоспоринам [24, 25]. В 2010 г. антибиотик зарегистрирован FDA по показаниям – «Внебольничная бактериальная пневмония» (включая случаи заболевания, осложненные вторичной пневмококковой бактериемией) и «Острые бактериальные инфекции кожи и мягких тканей»1.
В основе антимикробного действия цефтаролина, как и любого другого β-лактама, лежит ингибирование пенициллинсвязывающих белков (ПСБ) бактериальной стенки, что обусловливает нарушение ее новообразования и как следствие – лизис микроба. Установлено, что ПСБ у различных видов микроорганизмов демонстрируют известную структурную и функциональную гетерогенность [26]. В связи с этим важно подчеркнуть, что цефтаролин характеризуется высокой степенью сродства к ПСБ 2а, ответственных за развитие устойчивости S. aureus к метициллину (MRSA), равно как и к ПСБ 1a, 2b, 2x и 3, определяющих чувствительность к антибиотикам метициллиночувствительного S. aureus (MSSA) и S. pneumoniae [27]. При этом аффинность нового цефалоспорина к указанным ПСБ превосходит таковую оксациллина и цефтриаксона, являющихся общепринятым стандартом терапии MSSA- и S. pneumoniae-инфекций соответственно.
Очевидно, что одной из отличительных характеристик цефтаролина по сравнению с другими β-лактамами является его антистафилококковая активность, включая и лекарственно устойчивые штаммы возбудителя. Так, при изучении 152 изолятов внебольничного MRSA установлено, что минимальная подавляющая концентрация антибиотика в отношении 90% изучаемых штаммов (МПК90) оказалась равной 0,5 мкг/мл, в 64 раза превосходя активность цефтриаксона [28]. Выполненные в последующем исследования in vitro и in vivo подтвердили активность цефтаролина и в отношении штаммов, не чувствительных к ванкомицину и даптомицину [29, 30].
Однако очевидно, что применительно к лечению больных ВП особое значение приобретает активность цефтаролина в отношении ведущего возбудителя заболевания – S. pneumoniae, в т.ч. и его лекарственно-устойчивых штаммов. Важно подчеркнуть, в частности, что активность цефтаролина распространяется не только на пенициллиночувствительные пневмококки, но и на промежуточно устойчивые и устойчивые штаммы, в отношении которых МПК90 антибиотика составляла 0,06 мкг/мл, 0,13 и 0,25 мкг/мл соответственно, существенно превзойдя антипневмококковый потенциал цефтриаксона [24]. Цефтаролин сохраняет также активность в отношении штаммов S. pneumoniae, устойчивых к амоксициллину, эритромицину, цефотаксиму [31, 32].
Для определения роли и места цефтаролина в современных схемах лечения пневмонии особое значение приобретает анализ результатов исследований FOCUS 1 (NCT00621505) и FOCUS 2 (NCT00509106)2 – базовых рандомизированных многоцентровых3 двойных слепых клинических исследований по доказательству терапевтической эквивалентности цефтаролина и цефтриаксона у 1240 взрослых госпитализированных больных нетяжелой ВП [33, 34]. В отсутствие достоверных различий в клинической эффективности между цефтаролином и цефтриаксоном (84,5 и 77,7% соответственно) очевидным оказалось превосходство цефтаролина в группе больных пневмококковой ВП (85,5 и 68,9% соответственно), в т.ч. осложненной вторичной бактериемией (78,9 и 66,7% соответственно) [35].
Особый интерес представляла оценка клинического ответа (достижение клинической стабильности или клинического улучшения) больных ВП на 4-й день лечения. Выбор данного временнόго интервала имеет несомненное практическое значение, поскольку отсутствие терапевтического эффекта в первые 96 часов указывает на необходимость пересмотра лечебной тактики и позволяет избегать неоправданного «затягивания» неадекватной АБТ. В противоположность этому обнаруженное в первые 4 дня клиническое улучшение делает возможным осуществление деэскалации АБТ с переходом на прием антибиотика с узким (целенаправленным) спектром антимикробного действия, проведение ступенчатой терапии и последующей выписки больного из стационара [36, 37]. При этом, очевидно, что аргументированное сокращение длительности госпитального этапа лечения обусловливает снижение прямых затрат на ведение больного.
Эти, а также ряд других рандомизированных клинических исследований (РКИ) [38, 39], свидетельствовавшие о привлекательной эффективности и приемлемом профиле безопасности цефтаролина, аргументировали его появление в качестве возможной терапевтической опции на страницах современных рекомендаций по ведению взрослых больных ВП (табл. 2).

Тетрациклины: омадациклин
Омадациклин является ведущим соединением нового подкласса тетрациклинов – аминометилциклинов. Подобно тетрациклинам, омадациклин действует путем ингибирования синтеза белка, но связывается с 70S рибосом бактериальной клетки с большей аффинностью, чем тетрациклин [41]. Он является антибиотиком широкого спектра действия с улучшенной активностью в отношении устойчивых к тетрациклину патогенов, в т.ч. чувствительных к метициллину S. aureus (MSSA) и MRSA, а также различных стрепто- и энтерококков. Омадациклин также активен в отношении многих клинически важных энтеробактерий и широкого спектра анаэробов, а также «атипичных» микроорганизмов – Legionella pneumophila, Mycoplasma pneumoniae и Chlamydophila pneumoniae [42]. Омадациклин обладает фармакокинетическими преимуществами, а именно формированием более высоких и стабильных концентраций в плазме крови и слизистой оболочке дыхательных путей по сравнению с тигециклином, что позволяет предположить, что он может быть перспективным антибактериальным средством для лечения ВП, вызываемой чувствительными к нему возбудителями [43].
В США омадациклин был одобрен в октябре 2018 г. для лечения бактериальной ВП, инфекций кожи и мягких тканей. Его принимают 1 раз в сутки, что дает преимущество по сравнению с миноциклином и доксициклином, и он доступен как в пероральной, так и внутривенной лекарственных формах [44].
Одобрение FDA применения омадациклина в лечении ВП было основано на результатах РКИ III фазы с участием 750 пациентов (OPTIC Trail), в котором сравнивали пероральную или внутривенную формы омадациклина с пероральным или внутривенно вводимым моксифлоксацином в лечении больных ВП [45]. Пациентам омадациклин назначали по 100 мг внутривенно 2 раза в сутки (две дозы), затем по 100 мг ежедневно внутривенно или 400 мг моксифлоксацина внутривенно 1 раз в сутки в течение 3 дней.
В обоих случаях существовала возможность перейти на пероральный прием сравниваемых антибиотиков.
Омадациклин продемонстрировал сопоставимую с моксифлоксацином клиническую эффективность (81,1 и 82,7% соответственно) [46]. К сожалению, в исследование не были включены больные тяжелой ВП (соответствующие классу V по шкале PORT – Pneumonia Outcomes Research Team) и больные, течение пневмонии у которых было осложнено септическим шоком. НЛР со стороны желудочно-кишечного тракта (ЖКТ) были более частыми и демонстративными при применении омадациклина [47].
Кетолиды: солитромицин
Солитромицин является первым макролидом нового поколения класса фторкетолидов [48]. Антибиотик связывается с участком 50S рибосомы чувствительных бактерий, который либо перекрывается, либо совпадает с соответствующими участками других макролидов и кетолидов [49]. Способность солитромицина связываться с тремя различными участками 50S рибосомы (в отличие от телитромицина, который связывается только с двумя из них) объясняет меньшую вероятность селекции к нему устойчивых бактерий [50]. Кроме того, солитромицин обладает выраженной активностью in vitro в отношении наиболее распространенных бактериальных возбудителей ВП, включая устойчивые к макролидам, пенициллину и фторхинолонам изоляты S. pneumoniae, а также Haemophilus influenzae и «атипичные» бактерии. Фармакодинамически солитромицин в 16 раз активнее азитромицина, кларитромицина и телитромицина в отношении грамположительных аэробов, «атипичных» и грамотрицательных возбудителей [49]. Он обладает хорошей пероральной биодоступностью (67%), на которую не влияют сопутствующий прием пищи, связывание с белками сыворотки крови 81%, а период полураспада составляет 8,5 часов, что позволяет назначать антибиотик 1 раз в сутки [51]. Метаболизм солитромицина преимущественно связан с с цитохромом Р-450 (CYP) 3A4, при этом бόльшая часть метаболита выводится с желчью [52]. Безопасность и эффективность солитромицина в лечении нетяжелой бактериальной ВП первоначально была оценена в РКИ II (NCT01168713В) и III фаз (NCT01968733, NCT01756339) [53]. Солитромицин продемонстрировал сопоставимую клиническую эффективность с препаратом сравнения моксифлоксацином в первые 72 часа лечения (первичная «конечная точка»), что удовлетворяет требованиям FDA, но оказался менее эффективным на 5–10-е сутки (как того требует EMA4). Практически идентичным оказался ранний клинический эффект (оценка эффективности в первые 72 часа лечения) солитромицина и моксифлоксацина (79,3 и 79,7% соответственно) еще в одном исследовании III фазы (SOLITAIRE-IV), где для лечения нетяжелой ВП применялась ступенчатая схема применения нового кетолида [54].
Следует отметить, что солитромицин вызывает большее число НЛР (34%), в основном в месте инфузии, по сравнению с моксифлоксацином (13%), хотя и не увеличивает интервал QT на электрокардиограмме. Тот факт, что по сравнению с химической структурой телитромицина солитромицин отличается строением своей боковой цепочки, объясняет отсутствие у последнего влияния на никотиновые ацетилхолиновые рецепторы. Это обстоятельство способно минимизировать риск развития таких НЛР, как нарушения зрения, обострение миастении, потеря сознания, гепатотоксичность, свойственные раннему кетолиду. Это потенциально может уменьшить расплывчатое зрение, обострение миастении, потерю сознания и, как сообщается, своеобразную печеночную недостаточность с кетолидами [55]. Тем не менее проблема гепатотоксичности солитромицина сохраняет свою актуальность, поскольку при его приеме у 5–10% пациентов наблюдается умеренное повышение уровня трансаминаз. Учитывая относительно небольшие размеры выборки пациентов, включенных в исследования по оценке эффективности и безопасности применения солитромицина, эксперты FDA считают, что это может недооценивать реальную угрозу развития НЛР при его более широком клиническом применении. В связи с этим 29 декабря 2016 г. FDA рекомендовало провести дальнейшие исследования, увеличив число подвергшихся воздействию солитромицина пациентов с 924 примерно до 12 тыс., чтобы при принятии решения об официальном утверждении антибиотика риск его гепатотоксичности был бы оценен более объективно [66].
Хинолоны: немоноксацин, делафлоксацин
Репутация хинолонов как высокоэффективных препаратов в лечении ВП у взрослых, сформированная и поддерживаемая вот уже на протяжении 20 с лишним лет левофлоксацином и моксифлоксацином, объясняет сохраняющийся интерес к разработке новых представителей этого класса антибиотиков.
Немоноксацин. Немоноксацин – это новый нефторированный хинолон, который нацелен на ДНК-гиразу и топоизомеразу-IV и обладает более широким профилем антимикробной активности и меньшим риском селекции антибиотикорезистентных бактерий резистентности по сравнению с другими фторхинолонами.
К нему чувствительны многочисленные виды грамположительных, грамотрицательных и «атипичных» бактерий, включая MRSA (МПК90=1 мкг/мл), в т.ч. и устойчивые к ванкомицину [57]. При приеме внутрь 500 мг немоноксацина максимальная концентрация препарата в плазме крови достигается уже через 1–2 часа, период полувыведения составляет 12,8–18,5 часов, а биодоступность абсолютна (≈100%). Кроме того, для немоноксацина характерен низкий уровень связывания с белками плазмы (16%) и отсутствие известного взаимодействия с изоферментами системы цитохрома Р-450, что обеспечивает его минимальное потенциальное взаимодействие с другими лекарственными средствами. Немоноксацин доступен как в пероральной, так и во внутривенной лекарственных формах. На сегодняшний день проведены исследования II и III фаз по оценке сравнительной эффективности и безопасности пероральных форм немоноксацина и левофлоксацина, но только в исследовании II фазы сравнивались внутривенные формы немоноксацина и моксифлоксацина [58–60]. Так, в частности, было показано, что в группе больных ВП пожилого/старческого возраста (≥70 лет) немоноксацин и левофлоксацин продемонстрировали близкие показатели клинической эффективности (≈94%) и приемлемый профиль безопасности (при приеме немоноксацина наиболее частыми НЛР были побочные эффекты со стороны ЖКТ и нервной системы). На Тайване немоноксацин в лекарственной форме для приема внутрь был одобрен в марте 2014 г. для лечения бактериальной ВП, а к концу 2016 г. антибиотик поступил на рынок Турции, материкового Китая, Латинской Америки и ряда других стран [8]. Он уже получил статус приоритетного обзора FDA в качестве QIDP5, как только будут доступны дальнейшие исследования фазы III, документирующие его безопасность и эффективность.
Делафлоксацин. Делафлоксацин, в настоящее время продаваемый в США под торговым названием Baxdela®, представляет собой фторхинолон с активностью в отношении различных грамположительных бактерий, включая MRSA и устойчивые к хинолонам S. aureus, а также устойчивые к хинолонам штаммы P. aeruginosa и Klebsiella pneumonia [61, 62]. Его уникальная химическая структура способствует лучшему проникновению через клеточную мембрану и повышению эффективности в кислой среде, характерной для большинства очагов инфекционного воспаления. Он обладает новым механизмом действия, который ингибирует репликацию бактериальной ДНК путем одновременного связывания как с топоизомеразой IV, так и с ДНК-гиразой в отличие от «старых» фторхинолонов, которые только ингибируют оба фермента. Двойное «нацеливание» на ДНК-гиразу и топоизомеразу-IV снижает вероятность развития резистентности, поскольку в этом случае требуется накопление множества мутаций, которые бы нивелировали влияние антибиотика на оба фермента. Эта особенность делафлоксацина может объяснять его активность в отношении изолятов MRSA, в т.ч. тех, которые содержат мутации в отдельных областях субъединиц фермента и называются областями, определяющими устойчивость к хинолонам [62].
При проведении исследований в США и Европе показано, что ингибирование всех европейских и 98% североамериканских изолятов пневмококка достигается при концентрациях делафлоксацина <0,03 мкг/мл [63]. Делафлоксацин также активен в отношении P. aeruginosa (МПК50/90=0.25/4 мкг/мл), что делает его сопоставимым с другими антипсевдомональными фторхинолонами), а также анаэробов, «атипичных» возбудителей инфекций дыхательных путей (легионелл, хламидий и микоплазм), и даже против Micobacterium tuberculosis [64]. Биодоступность делафлоксацина составляет 58% при пероральном приеме, и его можно дозировать независимо от приема пищи. Антибиотик характеризуется минимальной способностью связываться с белками плазмы (16%); период его полувыведения при однократном внутривенном введении 300 мг составляет 3,7 часа, а в случае повторного приема внутрь колеблется от 4,2 до 8,4 часа. Примечательно, что делафлоксацин характеризуется высоким легочным распределением – соотношение концентрации антибиотика в легочной ткани и плазме крови составляет 13:1 [65].
Многообещающими оказались результаты РКИ по оценке эффективности делафлоксацина в лечении ВП. Так, в ходе одного из РКИ II фазы, включившего 309 амбулаторных пациентов, страдавших ВП, делафлоксацин назначался внутрь однократно в различных дозировках (100 мг, 200 или 400 мг) в течение 7 дней. При этом полные клиническое и бактериологическое излечения достигаются у 87% больных. В РКИ III фазы (DEFINE-BCAP) изучалась сравнительная эффективность делафлоксацин моксифлоксацина и линезолида в лечении взрослых больных бактериальным ВП, включая и случаи заболевания, вызванные MRSA [66]. На сегодняшний день частота НЛР делафлоксацина, по-видимому, зависит от дозы антибиотика, причем наиболее распространенными являются диарея и тошнота; среди других упоминаются влияние на центральную нервную систему, эндокринные нарушения и повышенные активности трансаминаз. Однако при исследовании на здоровых добровольцах случаев клинически значимого удлинения интервала QT/QTc зарегистрировано не было [67].
В настоящее время FDA определила делафлоксацин как QIDP и антибиотик получил статус ускоренного отслеживания для одобрения по показанию «Внебольничная пневмония» после завершения текущих РКИ III фазы.
Плевромутилины: лефамулин
Относящиеся к новому классу антибиотики являются полусинтетическими производными плевромутилина – натурального продукта грибов Pleurotus mutilus (в настоящее время называемых Clitopilus scyphoides). Их действие объясняется способностью ингибировать синтез белка бактериальными клетками путем связывания с участком пептидилтрансферазы 23S РНК 50S рибосом. Ретапамулин был первым препаратом, одобренным FDA в 2006 г., для местного применения в лечении импетиго. Другой представитель этого класса антибиотиков – тиамулин, с успехом использовался в ветеринарии в странах Европы и Канаде [68]. Однако лефамулин является первым плевромутилином, разработанным для перорального или внутривенного применения у людей. Лефамулин реализует свое антибактериальное действие путем связывания с 50S бактериальной рибосомы в четырех отдельных участках высококонсервативного ядра пептидилтрансферазного центра. Этот уникальный механизм действия объясняет и отсутствие перекрестной устойчивости к большинству доступных в настоящее время антибиотиков [69]. Действительно, препарат обладает выраженной активностью in vitro в отношении микроорганизмов, устойчивых к β-лактамным антибиотикам, фторхинолонам, макролидам, тетрациклинам и ванкомицину. Лефамулин демонстрирует хорошее накопление в жидкости, выстилающей дыхательные пути и альвеолы, превосходящее концентрацию в плазме крови в 5,7 раза [70]. Антибиотик характеризуется высокой степенью связывания с белками плазмы (80%), его период полувыведения достигает 12 часов, а выводится он преимущественно через ЖКТ в неизменном виде (86%) [71]. Режим дозирования лефамулина предполагает прием препарата внутрь 2 раза в сутки, но однократное внутривенное введение. Фармакодинамически он обладает высокой антистафилококковой активностью с МПК90, которая в 4 раза ниже аналогичной оксациллина, ванкомицина, линезолида или цефтаролина для S. aureus (включая MRSA) и многих других распространенных возбудителей ВП [72, 73]. В двух РКИ III фазы осуществлялась оценка сравнительной эффективности и безопасность лефамулина и моксифлоксацином в лечении взрослых больных ВП нетяжелого течения (NCT02559310 и NCT02813694) [74, 75]. При этом была продемонстрирована сопоставимая клиническая эффективность сравниваемых препаратов. В ряду НЛР лефамулина фигурировали головные боли (7%), тошнота (7%) и диарея/рвота (4%) [76]. Внутривенные и пероральные лекарственные формы лефамулина в настоящее время находятся на этапе одобрения FDA по показанию «Внебольничная пневмония».
Заключение
Очевидно, что доступность новых антибиотиков открывает возможности для расширения эмпирической терапии больных ВП, вызываемой лекарственно устойчивыми возбудителями. Однако при этом необходимо убедиться в том, что новые антибиотики эффективны и хорошо переносятся пациентами, а также в том, что их применение целесообразно с точки зрения минимизации риска развития антибиотикорезистентности. Из новых антибактериальных препаратов в РФ уже доступен и включен в клинические рекомендации цефтаролин, что расширяет возможности терапии ВП при наличии факторов резистентности и пациентов с высоким риском неудачи стартовой терапии при ТВП. При этом наряду с поиском новых антибиотиков необходима и разработка других потенциально эффективных «инструментов» предотвращения и лечения ВП – вакцин, моноклональных антител, малых молекул, что позволит свести к минимуму или избежать возникновения устойчивости к антибактериальным препаратам.
