Введение
Опухоли гипофиза составляют до 25% всех опухолей головного мозга. Наиболее часто встречаются аденомы – доброкачественные опухоли из клеток аденогипофиза [3, 4]. Выделяют спорадические и семейные формы аденом гипофиза (АГ). Подавляющее большинство аденом являются спорадическими. Семейные формы (как изолированные, так и в рамках эндокринных синдромов) составляют около 5% от всех случаев АГ [1– 3]. Этиология спорадических аденом до сих пор остается малоизученной, тогда как за последние десятилетия наблюдается значительный прогресс в изучении наследственных синдромов с возникновением АГ [3, 5]. К наследственным синдромам, в рамках которых могут развиваться АГ, относят синдром множественных эндокринных неоплазий 1-го типа (МЭН-1), синдром множественных эндокринных неоплазий 4-го типа (МЭН-4), Карни-комплекс (Carney complex, CNC) и семейные изолированные АГ (Familiar Isolated Pituitary Adenomas, FIPA) [3].
Семейные изолированные АГ (FIPA) характеризуются наличием аденом у двух и более членов одной семьи в отсутствие других синдромов, ассоциированных с опухолями [2, 6]. Около 2,7% всех АГ возникают в рамках МЭН-1, приблизительно 2,5% – в рамках FIPA [3, 7].
Первое исследование семейных АГ, не ассоциированных ни с синдромом МЭН-1, ни с Карни-комплексом, было проведено в Бельгии в 2000 г. в Льежском университете. В этом исследовании были описаны 27 пациентов с АГ в семьях, не имевших других выявленных опухолей [3]. К 2013 г. в литературе было описано более 400 семей с FIPA [3, 8].
Пролактиномы встречаются в 40% случаев FIPA, и их характеристики схожи со спорадическими пролактиномами по половому распределению и возрасту на момент постановки диагноза и проценту микроаденом [9, 10].
Исследователями отмечено, что в семьях могут наблюдаться как аденомы одного клинического типа, так и подтипы с различными клиническими проявлениями, что привело к субклассификации FIPA на гомогенные и гетерогенные семьи соответственно [11, 12]. В гетерогенных семьях пролактиномы ведут себя более агрессивно, со значительно большим преобладанием супраселлярного роста и инвазии кавернозных синусов по сравнению со спорадическими пролактиномами [3, 9, 13].
Более 75% семей с FIPA не имеют мутаций в гене AIP или других известных предрасполагающих генах, и это показывает, что другие генетические факторы еще предстоит выявить [10, 14, 15]. Генеалогические исследования продемонстрировали аутосомно-доминантный тип наследования FIPA с неполной пенетрантностью [1, 2, 16].
Клинический случай
На консультацию впервые обратилась пациентка А. 21 лет с жалобами на нерегулярные менструации, частые головные боли.
Из анамнеза заболевания стало известно, что нерегулярные менструации беспокоят больную около 6–7 месяцев. Менархе с 12 лет. При обследовании около полугода назад в гормональном анализе крови выявлена гиперпролактинемия – 710 мкМЕ/мл (норма – 102– 496). Лечения по поводу повышенного уровня пролактина не получала.
Из анамнеза жизни: наследственность не отягощена. Аллергии на пищевые продукты и лекарственные препараты ранее не отмечалось. Оперативные вмешательства отрицает.
Объективно: рост – 162 см, масса тела – 49 кг. Индекс массы тела – 18,8 кг/м2. Состояние удовлетворительное. Кожные покровы чистые, бледно-розовые, суховатые. Телосложение астеническое. Подкожная клетчатка развита недостаточно, распределена равномерно. ЧСС – 78 в минуту. АД – 110/80 мм рт.ст. Живот мягкий, безболезненный. Стул регулярный, оформленный. Диурез адекватный. Щитовидная железа не увеличена, при пальпации умеренно эластичной консистенции, клинически эутиреоз. В позе Ромберга устойчива. Периферические лимфатические узлы не увеличены. На момент осмотра признаков надпочечниковой недостаточности нет. Половое развитие по женскому типу, менструации нерегулярные, скудные, 1 раз в 30–45 дней продолжительностью 2–3 дня, безболезненные. Галактореи нет.
При дополнительном обследовании в гормональном профиле сохранялась гиперпролактинемия: уровень пролактина в крови – 731,6 мкМЕ/мл (норма – 102–496), уровень мономерного пролактина – 613,6 мкМЕ/мл (норма до 229). При проведении МРТ головного мозга были выявлены признаки микроаденомы гипофиза с максимальным диаметром 5 мм, неоднородность структуры аденогипофиза. Назначена терапия агонистом дофаминовых рецепторов пролонгированного действия каберголином в дозе 0,125 мг 2 раза в неделю. На фоне лечения отмечалась положительная динамика в виде нормализации менструального цикла, снижения уровня пролактина и уменьшения размеров аденомы гипофиза по данным МРТ-исследований (табл. 1, 2). Однако на протяжении 2 лет наблюдения достичь стойкой компенсации заболевания не удалось, в связи с тем что на фоне стабилизации менструального цикла пациентка самостоятельно прекращала прием каберголина и вновь начинала его при появлении признаков дисменореи. Наиболее длительный период приема препарата составил 6 месяцев. Динамика показателей пролактина и других гормонов представлена в табл. 1.
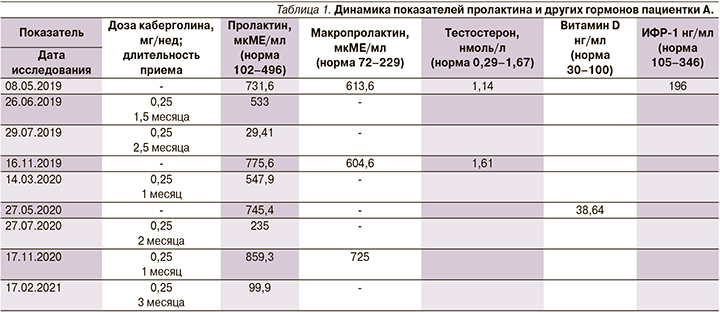

Спустя 2 года (февраль 2021 г.) с момента первичного обращения пациентки А. на прием обратилась ее мать Р. 47 лет со случайно выявленной гиперпролактинемией (1523,0 мкМЕ/мл) при диспансерном обследовании у гинеколога около 6 месяцев назад. Жалоб пациентка не предъявляла. Гинекологом была инициирована терапия каберголином в дозе 0,125 мг 2 раза в неделю без проведения МРТ-исследования головного мозга. На фоне регулярного приема каберголина в указанной дозе в течение 6 месяцев в гормональном профиле от 13.01.2021 сохранялась гиперпролактинемия: пролактин – 990,0 мкМЕ/мл (норма – 102–496), мономерный пролактин – 978 мкМЕ/мл (98,79%).
Объективно: рост – 172 см, масса тела – 68 кг. Индекс массы тела – 23,05 кг/м2. Состояние удовлетворительное. Кожные покровы чистые, бледно-розовые, умеренной влажности. Телосложение нормостеническое. Подкожная клетчатка развита умеренно, распределена равномерно. ЧСС – 81 в минуту. АД – 120/80 мм рт.ст. Живот мягкий, безболезненный. Стул регулярный, оформленный. Диурез адекватный. Щитовидная железа не увеличена, при пальпации умеренно эластичной консистенции, клинический эутиреоз. Периферические лимфатические узлы не увеличены.Признаков надпочечниковой недостаточности нет. Половое развитие по женскому типу, менструации регулярные, умеренные, 1 раз в 28–30 дней продолжительностью 4–5 дней, безболезненные. Галактореи нет.
По данным МРТ-исследования головного мозга от 15.02.2021 имеются признаки микроаденомы гипофиза с максимальным диаметром 4 мм.
В связи с отсутствием положительной динамики на протяжении 6 месяцев доза принимаемого каберголина была увеличена вдвое (до 0,25 мг) 2 раза в неделю с контролем уровня пролактина и макропролактина. Спустя месяц, несмотря на увеличение дозы каберголина вдвое, уровень пролактина у пациентки Р. повысился до 1225,0 мкМЕ/мл, что может свидетельствовать о частичной или полной резистентности пролактиномы к медикаментозному лечению.
С учетом того что пролактинома была диагностирована у членов одной семьи, нами было инициировано дополнительное обследование, невыявившее патологических отклонений со стороны других эндокринных желез и внутренних органов. Результаты обследования представлены в табл. 3.
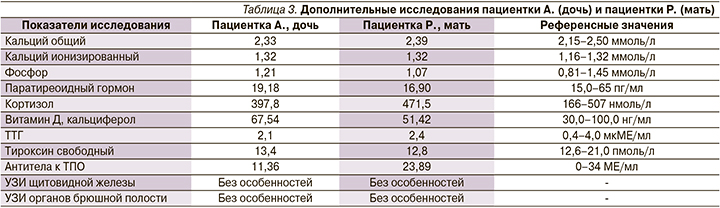
Таким образом, и дочери, и матери был установлен единый диагноз «семейная изолированная микроаденома гипофиза (пролактинома)».
Обе пациентки остаются под динамическим наблюдением и продолжают лечение агонистом дофаминовых рецепторов каберголином под контролем уровня пролактина и размеров АГ.
Обсуждение
С целью постановки окончательного диагноза нами была проведена дифференциальная диагностика с синдромами, в рамках которых встречаются АГ, в частности пролактиномы. Рассмотрим подробнее каждое из этих заболеваний.
За последние десятилетия к хорошо изученным синдромам МЭН-1 и Карни-комплекс (CNC) присоединились FIPA и МЭН 4, при которых также диагностируются пролактиномы. При этом велика вероятность существования других еще не открытых генов, вовлеченных в процесс образования АГ.
Синдром множественных эндокринных неоплазий 1-го типа. В большинстве случаев первым клиническим проявлением синдрома МЭН-1 является первичный гиперпаратиреоз, однако у 20% пациентов синдром манифестирует АГ [5]. Последние в рамках МЭН-1 возникают в более молодом возрасте (35,1±14,8 года) по сравнению с пациентами со спорадическими АГ [17]. Распределение типов АГ такое же, как и для не-МЭН-1-обусловленных (спорадических) аденом: преобладают пролактиномы (60%), менее 25% секретируют соматотропный гормон (СТГ), также встречаются гормонально неактивные аденомы (менее 5%), кортикотропиномы (менее 5%) и крайне редко тиреотропиномы [1, 3, 18]. От 10 до 39% АГ секретируют более одного гормона, обычно пролактин/СТГ. АГ в рамках МЭН-1 больше по размерам, чем спорадические аденомы: макроаденомы встречаются в 76–85% случаев при МЭН-1 по сравнению с 42% при не-МЭН-1 АГ, причем около половины макроаденом инвазивные [17]. У 4% пациентов развиваются множественные аденомы [1].
Ответ на терапию агонистами дофамина значительно меньше у МЭН-1-ассоциированных пролактином с нормализацией уровня пролактина в 42% случаев по сравнению с 90% в спорадических случаях [17]. На сегодняшний день проведено лишь небольшое количество исследований, посвященных проблеме лечения МЭН-1-ассоциированных АГ, тем не менее вероятно, что хирургическое лечение потребуется большему количеству пациентов с МЭН-1, чем в случае спорадических аденом [18].
Синдром множественных эндокринных неоплазий 4-го типа. Первым задокументированным случаем заболевания, в последующем названного МЭН-4, стала семья из Германии с семейными АГ, первичным гиперпаратиреозом, ангиомиолипомой почки и раком яичек среди различных членов семьи. Генетическое исследование показало наличие нонсенс-мутации в гене CDKN1B в отсутствие мутаций в гене MEN1 [3, 19]. Наиболее частым клиническим проявлением служит первичный гиперпаратиреоз, диагностированный у 81% пациентов. На втором месте по распространенности среди данных пациентов находятся АГ, наблюдавшиеся у 5 (41,6%) из 12 пациентов [20]. Поскольку количество выявленных пациентов крайне мало, пока невозможно достоверно утверждать, поражают ли мутации CDKN1B в равной степени все клетки аденогипофиза или являются специфичными лишь для определенных типов клеток. Также нельзя с уверенностью утверждать, являются ли АГ при наличии мутации CDKN1B более агрессивными по сравнению со спорадическими формами [3].
Карни-комплекс (Carney complex, CNC) – редкий наследственный синдром с аутосомно-доминантным типом наследования, характеризующийся множественными неоплазиями, в т.ч. органов эндокринной системы. К типичным проявлениям этого синдрома относятся микронодулярная пигментная дисплазия надпочечников, лентигиноз, миксомы сердца и кожи, крупноклеточные сертолиомы, парадоксальный ответ СТГ на введение тиротропин-рилизинг-гормона (ТРГ) или оральный глюкозотолерантный тест (ОГТТ), гиперпролактинемия, а также ряд других неоплазий. На сегодняшний день в мире описано всего несколько сотен таких пациентов [21].
Диагноз устанавливается на основании минимум двух главных критериев или наличия одного критерия и либо инактивирующей мутации в гене PRKAR1A, либо родственника первой линии с Карни-комплексом [22, 23].
АГ при Карни-комплексе в подавляющем большинстве случаев СТГ или СТГ/пролактинсекретирующие, около 2/3 пациентов с CNC имеют «мягкую» гиперпролактинемию, но описано нескольких случаев истинных пролактином [24]. Для пациентов с Карни-комплексом не характерно значительное повышение уровня пролактина, и встречается вследствие АГ относительно редко – в среднем у 10–12% пациентов [2].
Заключение
Спорадические пролактиномы являются наиболее распространенными из гормонально активных АГ. Их прогноз достаточно благоприятен, и в настоящее время разработан четкий алгоритм ведения пациентов с данной нозологией. При выявлении пролактиномы у членов одной семьи рекомендуется проведение дифференциальной диагностики АГ в рамках наследственных синдромов. Описанный клинический случай демонстрирует диагностику пролактиномы как семейной изолированной АГ в отсутствие признаков других заболеваний, характерных для МЭН-1, МЭН-4 и Карни-комплекса. Мутации в гене AIP, характерные для FIPA, наблюдаются только в 15–25% семей с FIPA, и их поиск не всегда помогает установить диагноз.
Согласие пациента. Пациентами добровольно подписано информированное согласие на публикацию персональной медицинской информации.
