Введение
Группа Т-клеточных лимфом кожи (ТКЛК) – это гетерогенная группа неходжкинских лимфом, которые представляют собой злокачественную трансформацию резидентных Т-лимфоцитов кожи [1]. Грибовидный микоз (ГМ) является наиболее частой формой ТКЛК и составляет почти 50% в структуре всех ТКЛК [2]. Опухоль состоит из малых/средних Т-лимфоцитов, характеризующихся тропностью к эпидермису (эпидермотропизмом). По определению, это заболевание имеет стадийное течение и характеризуется эволюцией сыпи – от пятен до бляшек и опухолей [2]. Как правило, ГМ поражает лиц взрослого и пожилого возраста (медиана – 55–60 лет), но может проявляться также у детей и подростков. Мужчины болеют несколько чаще женщин (1,5–2:1) [2].
Имеются сведения о дисрегуляции некоторых генов и их сигнальных путей при ТКЛК, но точно их роль в патогенезе заболевания неизвестна [2]. Нарушение экспрессии или функции негативных регуляторов, включая SOCS3 и протеинтирозинфосфатазы, такие как SHP1, вовлечено в дисрегуляцию пути Jak-3/STAT и интерлейкиннезависимой пролиферации злокачественных Т-клеток [2]. Активность пути Jak-3/STAT препятствует развитию ТКЛК через стимуляцию синтеза интерлейкина-5 (ИЛ-5), -10, -17A и -17F, регулированию факторов ангиогенеза и препятствует резистентности к терапии ингибитором гистоновой дезацетилазы (HDACI – histone deacetylase inhibitor) [2]. Дополнительно сообщается о вовлечении в патогенез синдрома Сезари сигнального пути NOTCH1. NOTCH включает семейство трансмембранных рецепторов, которые регулируют транскрипцию генов, участвующих в эмбриональном развитии, гомеостазе тканей, дифференцировке клеток и их выживаемости [2].
Течение заболевания достаточно индолентное, может длиться годами и даже десятилетиями [3]. До установления диагноза пациенты, как правило, имеют историю неспецифического экзематозного или псориазоформного дерматита с неспецифическими гистологическими характеристиками [3]. Медиана времени от первых клинических проявлений до установления диагноза составляет 4–6 лет, но может варьироваться от нескольких месяцев до 50 лет [3].
Начальные проявления ГМ характеризуются наличием различных по размеру эритематозных пятен с мелкопластинчатым шелушением, возможен незначительный зуд. Эти поражения могут иметь легкую степень атрофии и изменения, характерные для пойкилодермии [4]. Как правило, сыпь располагается на ягодицах, туловище и конечностях. С прогрессированием заболевания пятна превращаются в бляшки, имеющие насыщенный красно-коричневый цвет, анулярную, полицентрическую или подковообразную формы и пластинчатое шелушение на поверхности [4]. При крупноклеточной трансформации ГМ характерно появление язв на поверхности опухолей. Риск поражения других органов коррелирует со стадией заболевания и выше у пациентов с эритродермией или наличием опухолей [4]. Внекожная диссеминация начинается с регионарных лимфатических узлов, дренирующих наиболее пораженные участки кожи. Поражение костного мозга не характерно для ГМ и происходит нечасто [4].
Начальные проявления заболевания характеризуются наличием в поверхностных отделах дермы плотного лихеноидного лимфоцитарного инфильтрата с признаками эпидермотропизма и формированием в эпидермисе небольших скоплений клеток с типичными «церебриформными» ядрами (микроабсцессы Потрие) [5]. Опухолевые лимфоциты, которые содержатся в эпидермисе, чуть больше клеток дермального инфильтрата и имеют вокруг себя светлое гало. Вдоль дермоэпидермального соединения может наблюдаться формирование цепочек опухолевых клеток [5]. В эпидермисе может определяться акантоз, при этом спонгиоз, как правило, не наблюдается [6]. При прогрессировании заболевания инфильтрат может распространяться на все слои дермы с поражением подкожной жировой клетчатки, эпидермотропизм становится менее выраженным или даже исчезает [6]. Крупноклеточная трансформация ГМ характеризуется появлением крупных атипичных клеток в опухолевом инфильтрате, экспрессирующих на своей поверхности CD30 (около 25% клеток опухоли). Этот феномен ассоциируется с плохим течением заболевания [6]. Опухолевые клетки имеют фенотип α/β Т-хелперов памяти (βF1+, TCRγ-, CD3+, CD4+, CD5+, CD45RO+, CD8-, TIA-) [6]. В редких случаях может наблюдаться фенотип цитотоксических Т-лимфоцитов (βF1+, TCRγ-, CD3+, CD4-, CD5+, CD45RO+, CD8+, TIA+) или γ/δ Т-лимфоцитов (βF1-, TCRγ+, CD3+, CD4-, CD5+, CD45RO+, CD8+, TIA+), что не влияет на клиническое течение или прогноз заболевания [6]. В этих случаях корреляция с клинической картиной крайне важна для исключения агрессивных цитотоксических лимфом, таких как первичная агрессивная эпидермотропная CD8+-Т-клеточная лимфома или γ/δ-Т-клеточная лимфома кожи [6]. Потеря экспрессии пан-Т-клеточных маркеров (таких, как CD7) служит полезным диагностическим критерием, но этот феномен редко наблюдается на начальных стадиях заболевания, поэтому его диагностическое значение не следует переоценивать. Следует отметить, что при некоторых хронических дерматитах может наблюдаться потеря пан-Т-клеточных маркеров [6].
Дифференциальную диагностику ГМ проводят со следующими заболеваниями:
- Хронические дерматозы, которые могут иметь схожее клиническое течение (экзема, псориаз, реакции на введение лекарственных средств и т.п.).
- Хронические дерматозы, которые могут иметь подобные морфологические характеристики (лимфоматоидный контактный дерматит, лимфоматоидные реакции на введение лекарственных средств, актинический ретикулоид).
- Другие эпидермотропные ТКЛК (агрессивная эпидермотропная CD8+-Т-клеточная лимфома, γ/δ Т-клеточная лимфома и т.д.) [7].
Клинический случай
Пациентка Т. 60 лет в течение последнего года наблюдалась в отделении дерматовенерологии и косметологии ФГБУ «Поликлиника № 1» Управления делами президента с жалобами на распространенные зудящие высыпания на коже тела. Из анамнеза известно, что в течение 4 лет у пациентки наблюдается увеличение лимфатических узлов, по данным позитронно-эмиссионной компьютерной томографии 4 года назад заподозрена лимфома, после чего была проведена пункция правого подмышечного узла в НМИЦ гематологии, диагноз лимфомы был опровергнут, в связи с чем наблюдалась у онколога поликлиники с диагнозом «лимфаденопатия неясного генеза». Тогда же пациентка перенесла ветряную оспу, по поводу которой получала сопутствующее лечение у инфекциониста поликлиники. Три года назад впервые отметила появление высыпаний геморрагического характера на коже голеней, предплечий, тогда же был диагностирован сиалоденит. Была обследована дерматологом и ревматологом поликлиники, был поставлен диагноз «васкулит, органиченный кожей, неуточненный, вероятно вторичного генеза. Лимфоденопатия», Ревматологом был исключен системный ревматологический процесс. Учитывая лимфаденопатию, лимфоцитоз крови (лимфоциты 40%), данные за сиалоденит, для исключения лимфопролиферативного заболевания пациентка была направлена на консультацию гематолога, исключившего диагноз, а также для дифференциальной диагностики – на консультацию к инфекционисту. Пациентка была дообследована у инфекциониста, был поставлен диагноз: «ВЭБ (вирус Эпштейна–Барр)-инфекция, персистирующая форма, генерализованная лимфаденопатия». Ввиду отсутствия признаков активности ВЭБ-инфекции противовирусная терапия не проводилась. Тогда же пациентка была обследована на базе ГНЦДК, по данным гистологического исследования был подтвержден диагноз васкулита кожи.
Кожный процесс в течение последнего года был представлен мелкими папулами и макулами до 3 мм в диаметре, бледно-розового цвета на коже тела, геморрагической сыпью в области предплечий, голеней. В связи с этим в течение года пациентка наблюдалась у аллерголога и дерматолога поликлиники по поводу токсикодермии, получала инъекционные глюкокортикостероиды (ГКС) и антигистаминные препараты, инфузии ГКС и десенсибилизирующих препаратов, внутрь – антигистаминные препараты, наружно – топические ГКС и эмоленты с кратковременным положительным эффектом, быстро отмечала исчезновение высыпаний и их появление вновь. Параллельно была обследована гематологом поликлиники, в общем анализе крови: лимфоциты – 43%. Был поставлен диагноз «относительный лимфоцитоз реактивного генеза. Данных за системное заболевание кроветворной ткани нет». Также была обследована инфекционистом поликлиники: аутоантитела (АТ) к бореллиям и иерсиниям не обнаружены.
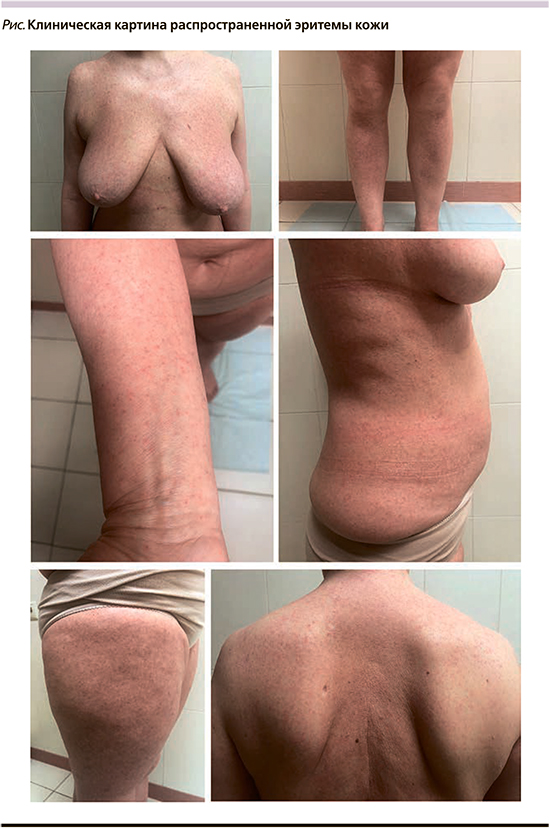
В течение последних месяцев отмечает сокращение периодов ремиссии, увеличение периодов обострения кожных процессов. Объективно наблюдается сохранение прежних элементов сыпи, появление сухости и распространенной эритемы кожи (см. рисунок). Пациентка повторно была обследована ревматологом поликлиники: АТ к двуспиральной ДНК, к ядерным антигенам (ANA) не обнаружены; С-реактивный белок норма, в общем анализе крови моноциты 15, нейтрофилы 33%, лимфоциты 46%. В связи с ухудшением кожного процесса, наличием стойких полиморфных высыпаний на теле, плохо реагирующих на проводимую системную и наружную терапию, пациентка вновь консультирована дерматологом и гематологом поликлиники. Был поставлен диагноз «лимфома кожи? Парапсориаз? Генерализованный васкулит кожи?». Была рекомендована повторная биопсия кожи для уточнения диагноза. Диагноз ГМ установлен по результатам гистологического и иммуногистохимического (ИГХ) исследований (РДБК им. Н.И. Пирогова). В гистологическом исследовании: в толще определялись немногочисленные лимфоидные скопления в виде абсцессов Потрие; субэпидермально – небольшой линейный инфильтрат из мелких лимфоцитов с признаками эпидермотропизма. При ИГХ: клетки в эпидермисе CD3/CD2/CD7/CD4 позитивные; субэпидермальные клетки экспрессируют CD3/CD5, фокально CD8. В настоящее время пациентка проходит дообследование и лечение в условиях НМИЦ гематологии.
Заключение
Данный клинический случай иллюстрирует сложность диагностики злокачественных лимфом кожи, в частности ГМ у пациентов ввиду различных его клинических вариаций, которые могут имитировать другие кожные заболевания, как в данном случае, когда пациентка длительно наблюдалась у дерматолога и аллерголога по поводу токсикодермии.
Таким образом, роль смежных специалистов в постановке диагноза остается неоспоримо высокой, а диагноз ГМ должен устанавливаться на основании комплексной оценки клинической картины, гистологического и иммунофенотипического исследований.
Согласие пациента. Пациентка добровольно подписала информированное согласие на публикацию персональной медицинской информации.
