Введение
Сахарный диабет (СД) – острейшая медико-социальная проблема современности, касающаяся многих людей, поскольку рост заболеваемости позволил говорить о глобальной эпидемии диабета. По данным Международной федерации диабета (IDF), на 2017 г. в мире насчитывают 425 млн больных СД [1]. На долю СД 2 типа (СД2) приходится 90% случаев заболевания. При недостаточно эффективном лечении СД2 может значительно ограничивать жизнь пациентов, приводить к ранней инвалидизации и преждевременной смертности из-за развития сосудистых осложнений, которые и определяют тяжесть недуга и ущерб, причиняемый обществу. Последствия СД обусловливают огромные экономические затраты для систем здравоохранения, поскольку около 80% затрат на лечение СД связано именно с терапией сосудистых осложнений. Так, присоединение диабетических осложнений в среднем удорожает лечение в 3–10 раз [2].
Среди патогенетических механизмов развития гипергликемии инсулинорезистентность (ИР) играет ключевую роль, поэтому неудивительно, что успехи, достигнутые в лечении СД, в значительной мере связаны с использованием лекарственных средств, улучшающих чувствительность к инсулину [3]. Установлена существенная роль ИР в преждевременном развитии сердечно-сосудистых заболеваний (ССЗ), связанных с атеросклерозом, а также в повышении риска острых макроваскулярных осложнений вне зависимости от других значимых факторов риска, включая гипергликемию, дислипидемию, курение [4, 5].
Выбор ИР, ключевого патофизиологического звена СД2, в качестве стратегической мишени лечения позволяет улучшать чувствительность к инсулину и позитивно воздействовать на сопутствующие метаболические нарушения. Фармакологическая коррекция ИР увеличивает эффективность сахароснижающей терапии, направленной на другие патофизиологические звенья развития СД2 [6–8]. Выраженным ингибирующим воздействием на ИР обладают бигуаниды. История их применения началась еще в средние века с применения гуанидина, который получали из растения Galega officinalis (галега лекарственная, французская лилия). Однако выраженная токсичность препятствовала клиническому применению этого вещества [9].
В настоящее время метформин является единственным бигуанидом, рекомендованным для фармакотерапии больных СД2. Благодаря высокой клинической эффективности, уникальным терапевтическим свойствам, хорошему профилю безопасности, доказанными в клинических исследованиях и подтвержденными многолетним опытом применения, в современных клинических рекомендациях метформин закрепил ведущую позицию в лечении СД2 (при условии отсутствия противопоказаний к нему) и применяется на всех этапах течения заболевания [8, 10, 11]. Впервые клинические эффекты метформина были представлены после завершения многолетнего исследования UKPDS (United Kingdom Prospective Diabetes Study) в 1998 г. [12]: снижение риска сосудистых осложнений на 32%, инфаркта миокарда на 39%, смертности от диабета на 42% и общей смертности на 36%. С 2005 г. метформин считается препаратом первой линии фармакологического лечения СД2 в комплексе с нефармакологическими методами, согласно рекомендациям IDF, с 2006 г. – согласно рекомендациям Американской диабетической ассоциации (American Diabetes Association – ADA) и Европейской ассоциации по изучению диабета (European Association for the Study of Diabetes – EASD) [13].
Механизм действия и сахароснижающий (антигипергликемический) эффект метформина
Антигипергликемические эффекты метформина – это результат воздействия препарата на тканевую чувствительность к инсулину. Хотя преобладающим является влияние метформина на гепатическую продукцию глюкозы, именно комбинация эффектов на уровне печени, мышечной и жировой тканей в целом обеспечивает благоприятный фармакологический профиль [6, 7, 9]. Известно, что последствия избыточной эндогенной продукции глюкозы печенью особенно неблагоприятны ввиду стимуляции процессов атерогенеза и развития резистентности к действию сахароснижающих препаратов в течение дня. В частности, при повышении концентрации глюкозы крови натощак (ГКН) >6,1 ммоль/л риск развития сердечно-сосудистых событий в последующие 12,4 года возрастает в 1,33 раза [14]. Применение метформина приводит к снижению на 25–30% уровня ГКН, постпрандиальной гликемии (табл. 1) [8, 13].

За антитипергликемический эффект метформина ответственны несколько механизмов, причем основное его действие осуществляется на уровне митохондрий гепатоцитов [9, 13]. Молекулярной мишенью действия метформина служит аденозинмонофосфат-активируемая протеинкиназа (АМФК), известная как ключевой фермент клеточного метаболизма и энергетического баланса (см. рисунок). Опосредованная метформином активация АМФК приводит к восстановлению энергетического гомеостаза путем увеличения поглощения глюкозы в скелетных мышцах и подавления глюконеогенеза в печени [15, 16]. Под воздействием метформина активируется сигнальный путь печеночной киназы В1(LKB1)/AMФK; в котором LKB1 является восходящим активатором AMФK, отвечающим за фосфорилирование Thr 172 каталитической α-субъединицы этой киназы. Активация АМФК вторична по отношению к влиянию метформина на митохондриальную дыхательную цепь. Как наиболее вероятный механизм, объясняющий активацию AMФK, рассматривается следующий. АМФК активируется путем фосфорилирования LKB1-киназы в ответ на повышение соотношения АМФ/АТФ внутри клетки посредством ингибирования митохондриальной дыхательной цепи и увеличения активных форм азота под влиянием метформина. Активация АМФК подавляет экспрессию генов, отвечающих за ферменты глюконеогенеза [15, 17].

Усиливаются эффекты инсулина на транскрипцию, трансляцию и синтез фосфатидилинозитол-3-киназы, ответственной за транслокацию переносчиков глюкозы к плазматической мембране, что приводит к увеличению поглощения глюкозы печеночными, мышечными и жировыми клетками [6, 8]. Параллельно с этим происходит окисление жирных кислот.
В мышечной и жировой ткани метформин повышает связывание инсулина с рецепторами, также наблюдается повышение их числа и аффинности. Кроме того, происходит активирование пострецепторных механизмов действия инсулина, в частности тирозинкиназы и фосфотирозин фосфатазы [7, 10].
Изучение клеточно-молекулярных механизмов, лежащих в основе действия метформина, показало, что ингибирование экспрессии генов глюконеогенеза не единственный механизм гепатических эффектов метформина (AMPK-независимые механизмы) [17]. Другая концепция основывается на тесной взаимосвязи энергетического профиля гепатоцитов и гепатической продукции глюкозы – процесса энергозатратного. На фоне снижения уровня АТФ, вызываемого метформином, гепатоцитам приходится, соответственно, уменьшать продукцию глюкозы [18].
Заслуживает обсуждения и еще один механизм антигипергликемического действия препарата. Метформин модулирует активность инкретиновой оси и повышает уровень глюкагоноподобного пептида-1 (ГПП-1) [17, 19, 20]. Существует несколько гипотез по поводу того, как метформин способствует секреции ГПП-1. В частности, метформин стимулирует секрецию ГПП-1 через активацию Wnt-сигнальных путей в L-клетках кишечника у мышей линии db/db. Кроме того, стимулирует экспрессию генов, кодирующих рецепторы ГПП-1 и глюкозозависимый инсулинотропный полипептид (ГИП) в островках Лангерганса, увеличивает экспрессию генов белков-предшественников – преглюкагона, проглюкагона [19]. Данные относительно влияния метформина на активность дипептидилпептидазы-4 (ДПП-4) противоречивы. В исследованиях in vivo в отличие от in vitro показано, что применение метформина ассоциируется со снижением уровня ДПП-4 (возможно, через влияние на гепатическую продукцию ДПП-4).
Есть мнение о непрямых действиях метформина на секрецию ГПП-1, которые опосредуются изменением энтерогепатической циркуляции желчных кислот. Метформин подавляет ядерные фарнезоидные Х-рецепторы (FXR) AMPK-зависимым механизмом. Выступая как АМРК-активатор, метформин подавляет транскрипционную активность FXR. В результате снижается кишечная абсорбция желчных кислот и возрастает их пул в подвздошной кишке. Желчные кислоты являются естественными лигандами мембранного рецептора желчных кислот или TGR5 энтерохромофинных клеток кишечника. В свою очередь стимуляция TGR5 индуцирует секрецию ГПП-1 L-клетками. Активация TGR5 увеличивает расход энергии и потребление кислорода, тем самым улучшая чувствительность к инсулину [21].
Метформин и функция β-клеток
СД2 считается хроническим прогрессирующим заболеванием, поэтому крайне важно сохранение функциональной активности β-клеточного аппарата поджелудочной железы [22]. Принципиально важно, что, повышая печеночную и периферическую чувствительность к инсулину, метформин не влияет на секрецию инсулина [6, 10, 17]. Вместе с тем повышая чувствительность тканей к действию инсулина, снижая глюко- и липотоксичность, препарат опосредованно улучшает секрецию инсулина и способствует сохранению функциональной активности β-клеток. Благодаря всем указанным эффектам метформина снижение уровня глюкозы происходит без риска гипогликемических состояний, что считается несомненным достоинством препарата [23].
В этом плане интересны результаты ретроспективного исследования S.A. Berkowitz и соавт., в котором была проанализирована сахароснижающая терапия на этапе ее инициации и интенсификации с оценкой данных более чем 15,5 тыс. пациентов [24]. Согласно дизайну, пациентов, которым впервые назначались пероральные сахароснижающие препараты (ПСП) в период с 2009 по 2013 г., классифицировали в зависимости от получаемого лечения на несколько групп: только метформин или препараты сульфонилмочевины (ПСМ), или тиазолидиндионы, или только ингибиторы ДПП-4.
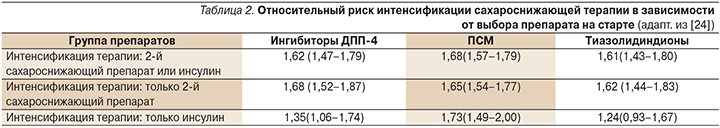
Целью данного исследования стало определить время интенсификации терапии (путем добавления другого сахароснижающего средства, включая инсулин) у пациентов с СД2, которым впервые были назначены ПСП. Как оказалось, несмотря на существующие алгоритмы лечения СД2, только 57,8% пациентов начинали терапию заболевания с приема метформина. С помощью регрессионного анализа Кокса в обширной популяции пациентов авторами показано, что старт терапии с метформина ассоциировался с меньшей необходимостью интенсификации терапии в будущем по сравнению с другими ПСП (табл. 2). В частности, доля пациентов, которым потребовалось назначение второго сахароснижающего средства и получавших на старте терапии метформин, составила 24,5%, ингибиторы ДПП-4 – 36,2%, ПСМ – 37,1%, тиазолидиндионы – 39,6%.
Снижая концентрацию и окисление свободных жирных кислот (СЖК) на 10–17 и 10–30% соответственно, активизируя их реэстерификацию, метформин не только улучшает тканевую чувствительность к инсулину, но и препятствует прогрессированию нарушений секреции инсулина у больных СД2 [7, 10, 13]. Последствия избыточной продукции СЖК сводятся к нарушению их компенсаторного окисления. Как следствие – к активации неокислительных путей метаболизма с образованием метаболитов, оказывающих липотоксическое действие на ткани (печень, сердце, мышечная ткань, поджелудочная железа). В частности, избыток СЖК подавляет активность пируватдегидрогеназы, нарушает транспорт и фосфорилирование глюкозы в скелетных мышцах. Повышенная концентрация СЖК на уровне печени стимулирует глюконеогенез. В свою очередь нормализация концентрации СЖК на фоне применения метформина приводит к устранению эффектов липотоксичности на всех уровнях, включая печень, жировую и мышечную ткани и островки Лангерганса. Снижение поступления СЖК к печени, синтеза триглицеридов и повышение чувствительности к инсулину сопровождаются уменьшением отложения жира в этом органе [17, 18].
Как известно, глюкозотоксичность служит ключевым фактором прогрессирования дисфункциb β-клеток [22]. Оказывая влияние на всасывание углеводов в желудочно-кишечном тракте, замедляя его скорость, а также снижая аппетит, метформин способствует уменьшению постпрандиальной гликемии, что также способствует устранению глюкозотоксичности. Кроме того, метформин повышает утилизацию глюкозы в кишечнике, усиливая в нем анаэробный гликолиз как в состоянии насыщения, так и натощак. В результате на фоне терапии метформином постпрандиальная гликемия снижается в среднем на 20–45% [17, 23].
Метформин и масса тела
Важной задачей лечения СД2 является снижение и поддержание нормальной массы тела. Согласно данным клинических исследований, терапия метформином препятствует увеличению массы тела либо способствует ее снижению, в т.ч. и в комбинации с инсулином. В зависимости от длительности приема препарата снижение массы тела у больных СД2 составляет от 0,5 до 6,0 кг [9, 10, 13]. Это остается значимым преимуществом препарата, поскольку применение ПСМ обычно вызывает увеличение массы тела. Интересный факт: терапия метформином сопровождается снижением висцерального отложения жира, которое является независимым фактором ССЗ [25].
Потенциальное влияние метформина на массу тела обусловлено несколькими механизмами, в т.ч. снижением концентрации инсулина, увеличением секреции ГПП-1. Согласно результатам экспериментальных исследований на животных моделях, метформину присущ анорексигенный эффект, по-видимому опосредованный центральным действием препарата на уровне гипоталамических нейронов [26]. Улучшение чувствительности к инсулину на фоне метформина устраняет причину для гиперинсулинемии, что также способствует снижению аппетита и уменьшению потребления энергии (в среднем – 104,5 ккал/сут) [27].
Применение метформина сопровождается подавлением выработки орексигенного пептида грелина, повышением концентрации ГПП-1, обладающего анорексигенным действием, что отчасти объясняет некоторые метаболические эффекты [17, 20, 25]. Кроме того, модулируя экспрессию орексигенного нейропептида Y, препарат способствует снижению массы тела [28].
Внегликемические эффекты метформина
В настоящее время немалое внимание уделяется внегликемическим эффектам сахароснижающей терапии, поскольку важно не только достигать контроля гликемии, но и воздействовать на другие модифицируемые факторы риска развития сердечно-сосудистой патологии для улучшения прогноза жизни.
Оценивая эффективность метформина, чаще обращаются к гликемическому контролю, однако немалое значение приобретают и иные эффекты (гиполипидемические, вазопротекторные и др.) препарата [6, 7, 10]. Примерно половина пациентов с СД2 имеют атерогенную дислипидемию, что неблагоприятно влияет на кардиоваскулярный прогноз. Эффекты метформина в отношении метаболизма липидов плазмы обусловлены его гиполипидемическим и антиатерогенным действиями. Как уже говорилось, метформин активирует АМФК, в результате подавляется экспрессия генов липидного метаболизма, таких как синтетазы СЖК (G001196), ацетил-CoA-карбоксилазы (G000684), S14, белка, связывающего стерол-регулирующий элемент – 1 (SREBP-1c) [29]. Помимо сказанного метформин влияет на процесс биосинтеза мононенасыщенных жирных кислот из насыщенных жирных кислот через АМФК-опосредованное фосфорилирование рецептора тиреоидных гормонов 4 (TR4) [17].
Кроме влияния на метаболизм СЖК лечение метформином ассоциируется с позитивными изменениями в липидном спектре; в частности снижением на 10–20% концентрации триглицеридов, на 10% – липопротеидов низкой плотности и повышением на 10–20% концентрации липопротеидов высокой плотности [13, 17, 23]. Подобные эффекты служат дополнительным преимуществом метформина с позиции основной цели терапии СД2 – снижение кардиоваскулярной заболеваемости и смертности.
Информация была бы неполной, если не привести результаты экспериментальных работ, свидетельствующие о влиянии метформина на метаболизм липидов сосудистой стенки. В частности, препарат снижает аккумуляцию эфиров холестерина в аорте, увеличивает содержание фосфолипидов и уменьшает содержание сфингомиелина. Наряду с этим метформин уменьшает пролиферацию гладкомышечных клеток (ГМК) сосудов. Снижая отложение липидов в сосудистой стенке, уменьшая пролиферацию ГМК, нарушая адгезию, трансформацию моноцитов и способность захватывать липиды, метформин активно воздействует на ранние стадии развития атеросклероза [30]. Следует отметить, что, подавляя повышенную адгезию моноцитов к эндотелию сосудов, липоидоз, метформин влияет на пусковые механизмы развития атеросклеротического поражения, снижает экспрессию рецепторов, вовлеченных в процессы внутриклеточного накопления липидов [31]. Кроме того, метформин подавляет процессы дифференциации моноцитов в макрофаги, активно секретирующие проатерогенные факторы. In vitro показано ингибирующее влияние метформина на лейкоцит-эндотелиальное взаимодействие, а также экспрессию на поверхности эндотелия таких молекул адгезии, как внутриклеточная молекула адгезии 1, сосудисто-клеточная молекула адгезии 1 и Е-селектин [10, 31].
Дополнительные кардиопротективные эффекты метформина обусловлены и его влиянием на систему гемостаза, реологию крови (антитромбическое действие), функцию эндотелия и сосудистую реактивность (табл. 3).

В частности, метформин снижает уровень маркеров активации тромбоцитов (тромбоцитарного фактора 4 и β-тромбоглобулина), концентрацию ингибитора активатора плазминогена-1, выработку молекул адгезии и фибриногена, тормозит активность XIII и VII факторов свертывания [8, 13, 30].
Вазопротективные свойства метформина многоплановые: нормализация цикла сокращение/расслабление артериол, уменьшение проницаемости сосудистой стенки и торможение процессов неоангиогенеза, улучшение эндотелийзависимой вазодилатации и транспорта глюкозы в эндотелиоцитах, ГМК сосудов, а также в миокарде [23, 32].
Метформин обладает прямым противовоспалительным действием на сосудистую стенку, что в основном обусловлено подавлением сигнальных путей образования NF-kB, активация которого приводит к инициации транскрипции генов ряда цитокинов и молекул адгезии [17].
В совокупности гликемические эффекты и дополнительные свойства (антисклеротические, вазопротективные, антивоспалительные, антиоксидантные) метформина важны для снижения риска возникновения ССЗ и увеличения продолжительности жизни пациентов с СД2. В настоящее время СД2 рассматривается как эквивалент наличия у больного клинически выраженного ССЗ, что выдвигает серьезные требования к долгосрочной кардиоваскулярной безопасности сахароснижающих препаратов [5]. Хорошая иллюстрация кардиопротективных свойств метформина – это результаты исследования UKPDS, касающиеся общих и сердечно-сосудистых исходов. Впервые благоприятное воздействие метформина на частоту развития ССЗ у больных СД и ожирением установлено в исследовании UKPDS [12]. В дальнейшем и другие исследования показали снижение сердечно-сосудистой и общей смертности среди больных СД2, получавших метформин в монотерапии или в комбинации с другими сахароснижающими препаратами [33–35]. В частности, результат Канадского ретроспективного анализа базы данных пациентов (Saskatchewan Health databases, n=12 272) показал значительное сокращение общей и сердечно-сосудистой смертности на 40 и 36% соответственно [33]. В исследовании PRESTO (Prevention of Restenosis with Tranilast and its Outcomes) применение метформина ассоциировалось с улучшением прогноза – снижением риска развития всех клинически значимых событий (28%), инфаркта миокарда (69%) и смертности от всех причин (61%) [34].
Недавно были опубликованы результаты ретроспективного анализа сердечно-сосудистой безопасности 12 020 тыс. пациентов с СД2 (the UK Clinical Practice Research Datalink), которым начиная с 2000 г. была назначена инсулинотерапия [35]. Первичной конечной точкой исследования была смертность от любой причины, вторичная конечная точка включала инфаркт миокарда и инсульт. В результате применение инсулина в комбинации с метформином ассоциировано с 40%-ным снижением риска смерти от всех причин по сравнению с монотерапией инсулином (отношение рисков – ОР=0,60, 95% доверительный интервал [ДИ] – 0,52–0,68). Среди пациентов, получавших инсулин в комбинации с метформином, по сравнению с пациентами на монотерапии инсулином (n=6484) отмечено снижение риска инфаркта миокарда, инсульта на 25% (ОР=0,75, 95% ДИ – 0,52–0,68).
В научных публикациях появляется все больше информации о новых мишенях метформина. Стало очевидным, что метаболические эффекты метформина отчасти связаны с изменением кишечной микробиоты [17, 20]. В частности, применение метформина сопровождается повышением количества видов бактерий рода Escherichia, Akkermansia muciniphila, Subdoligranulum, в т.ч. бактерий, продуцирующих короткоцепочечные жирные кислоты (бутират), которые стимулируют синтез инкретинов. Микробный анализ показал достоверное снижение потенциала микробиоты к производству бутирата у больных СД2 [33, 37].
Другие области применения метформина
Результаты многочисленных экспериментальных и клинических исследований позволили раскрыть новые аспекты действия метформина, полнее оценить его практическую значимость, а также расширить показания к его назначению [15, 17, 18, 20].
В последние годы большое внимание уделяется антиканцерогенному действию метформина, которое скорее всего осуществляется через активацию АМФК. Это приводит к ингибированию активности M-TOR (mammalian target of rapamycin), центрального регулятора синтеза белка и клеточного роста, с последующим восстановлением чувствительности к инсулину и снижением гиперинсулинемии [17, 39]. Эффект снижения уровня инсулина метформином может играть важную роль в противоопухолевой активности, т.к. инсулин обладает митогенными и пролиферативными свойствами и опухолевые клетки часто экспрессируют высокие уровни рецептора инсулина [38–40]. Метформин является также эффективным в лечении синдрома поликистозных яичников [41].
Практические аспекты применения метформина
Благодаря отсутствию длинных гидрофобных боковых цепей ограничивается как способность препарата к связыванию с клеточной мембраной, так и активное накопление внутри клетки, что определяет низкую вероятность лактат-ацидоза и высокую безопасность терапии метформином [6, 9, 10]. Лечение препаратом инициируют с 500–850 мг, принимаемыми за ужином или на ночь. Во избежание побочных эффектов метформина (диспепсические явления, диарея, абдоминальный дискомфорт) необходима постепенная титрация дозы препарата, увеличение на 500–850 мг каждые 1–2 недели, а в некоторых случаях временное снижение до предыдущей дозы. Побочные эффекты обычно исчезают при снижении дозы препарата. Максимальная рекомендованная доза больным метаболическим синдромом составляет 2500–3000 мг/сут в 2–3 приема.
Прием метформина может быть продолжен до скорости клубочковой фильтрации >45 мл/мин/1,73 м² в отсутствие других противопоказаний:
- заболевания, сопровождающиеся тканевой гипоксией (сердечная или легочная недостаточность, инфаркт миокарда, анемия и др.);
- почечная недостаточность или нарушение функции почек (клиренс креатинина <45 мл/мин/1,73 м²);
- печеночная недостаточность, алкоголизм;
- беременность, лактация;
- острые состояния, которые могут нарушать функцию почек (обезвоживание, острая инфекция, шок, внутрисосудистое введение рентгеноконтрастных средств);
- диабетический кетоацидоз.
Следует воздержаться от назначения препарата в период беременности и лактации.
Результаты клинических исследований свидетельствуют, что метформин снижает уровень гликированного гемоглобина (НbА1с) на 0,6–2,4% [6–9]. Такая вариабельность степени снижения гликемии связана с исходной концентрацией глюкозы в крови до инициации лечения этим препаратом. Отсутствие достижения целевого уровня НbА1с на фоне монотерапии метформином свидетельствует о значительном нарушении секреторной активности β-клеток. В этих случаях необходимо применение комбинации метформина с сахароснижающим препаратом иного механизма действия. Механизм действия метформина позволяет успешно комбинировать метформин со всеми сахароснижающими препаратами (сульфонилмочевиной, меглитинидами, тиазолидиндионами, агонистами ГПП1, ингибиторами ДПП4), что повышает общую терапевтическую эффективность и улучшает гликемический контроль [8, 18, 23]. Отдельно следует отметить, что рациональна комбинация метформина и инсулина, при таком варианте значительно улучшается чувствительность тканей и к экзогенному инсулину. В результате суточная доза инсулина снижается в среднем на 17–30%, уменьшается риск гипогликемий, не увеличивается риск прибавки массы тела [13].
Заключение
Очевидно, что СД2 представляет серьезную проблему здоровья населения Российской Федерации. Решение этой проблемы требует повышения эффективности сахароснижающей фармакотерапии, в которой большое значение имеет применение лекарственных средств, воздействующих на ИР. Метформин – высокоэффективный и безопасный сахароснижающий препарат с обширной доказательной базой и огромным опытом применения в условиях российской клинической практики.
