Введение
В настоящее время известно более 70 лизосомных болезней накопления (ЛБН) – редких заболеваний, при которых дефект ферментов, обусловленный генетическими изменениями, вызывает накопление в лизосомах продуктов разрушения клеток. Среди ЛБН выделяют мукополисахаридоз I типа, который, как и другие мукополисахаридозы, считается клинически неоднородным заболеванием с точки зрения распространенности и скорости прогрессирования поражения органов. Такие методы лечения, как трансплантация гемопоэтических стволовых клеток и заместительная ферментная терапия, дают возможность значительно улучшать прогноз при наличии этого хронического жизнеугрожающего наследственного заболевания. Рано поставленный диагноз и вовремя назначенное лечение могут предотвращать необратимый ущерб здоровью пациента.
Мукополисахаридоз I типа (МПС-I) – наследственная болезнь накопления, вызванная недостаточностью лизосомального фермента α-L-идуронидазы, которая приводит к прогрессирующему накоплению глюкозаминогликанов (ГАГ) в разных тканях и органах [1]. МПС-I является аутосомно-рецессивной патологией, развивающейся вследствие мутации в гене IDUA в хромосомной области 4p16.3 [2, 3]. Болезнь характеризуется широким спектром клинических симптомов. В настоящее время различают три фенотипа болезни: синдром Гурлер – тяжелая форма, синдром Шейе – легкая, а также синдром Гурлер–Шейе – среднетяжелая форма [4–8].
Ранние диагностика и лечение имеют ключевое значение для прогноза жизни пациентов с МПС-I.
У пациентов с синдромом Гурлер с первых лет жизни обнаруживаются грубые черты лица, умственная отсталость, помутнение роговицы, тугоподвижность суставов. Одно из ведущих проявлений заболевания – тяжелое поражение центральной нервной системы, что приводит к ранней инвалидизации и летальному исходу в первые десять лет жизни при присоединении патологии дыхательной и сердечно-сосудистой систем [9–11]. В дебюте болезни у пациентов наблюдаются риниты, отиты, паховые и пупочные грыжи, однако эти симптомы могут встречаться и у младенцев без МПС-I. Поэтому у пациентов с легкой степенью выраженности заболевания ранняя идентификация болезни затруднительна. И именно такие пациенты, несмотря на четко отработанные критерии генетической диагностики, представляют серьезную проблему при постановке диагноза в результате «смазанной» первичной клинической картины. По данным зарубежных авторов, пациенты с поражением суставов, проявляющимся тугоподвижностью и последующим развитием контрактур, часто попадают к детским ревматологам или наблюдаются у педиатров по поводу некоего ревматологического заболевания без правильно установленного диагноза МПС, что ухудшает прогноз заболевания и лишает пациентов возможности вовремя получать необходимую терапию [12–16].
Поскольку МПС-I относится к редким наследственным заболеваниям, это исключает возможность проведения больших когортных эпидемиологических и рандомизированных контролируемых исследований и препятствует созданию эффективных протоколов диагностики и терапии. Важно научить врачей-педиатров распознавать заболевание уже на основе собранных анамнестических данных. Критерии диагностики можно сформировать только на основании интеграции скрининговых программ и накопленного опыта.
В данной статье приводятся результаты пилотного проекта, целью которого является формирование критериев выявления МПС-1 в рутинной клинической практике педиатра. Формированию критериев способствовали интеграция программы молекулярно-генетического скрининга заболевания на территории Российской Федерации (РФ), а также скрининговое исследование медицинских карт пациентов с установленным диагнозом и подозрением на МПС-I.
Материал и методы
На основании данных литературы была составлена анкета, включающая сбор параметров, характеризующих состояние органов и систем организма пациента с МПС-1, а именно: нарушения со стороны опорно-двигательной системы, желудочно-кишечного тракта, сердечно-сосудистой системы, нервной системы, офтальмологические и оториноларингологические нарушения, а также внешние фенотипические признаки и результаты энзимодиагностики.
В очном анкетировании приняли участие 16 пациентов* (в возрасте от 3 месяцев до 15 лет) с генетически подтвержденным диагнозом МПС-I, которые проходили обследование в НМИЦ здоровья детей Минздрава России, и 59 пациентов (в возрасте от 3 месяцев до 16,7 года) с подозрением на МПС, обследованных в том же учреждении и в Клиническом центре им. И.М. Сеченова, которым проведена энзимодиагностика на МПС-I, однако диагноз не подтвердился.
Кроме того, для статистического анализа были использованы результаты продиагностированных в 2017 г. (январь–август) 738 пациентов из разных регионов РФ, из которых у 7 пациентов был подтвержден диагноз МПС-I.
Для создания валидных групп критериев, способствующих дифференциальной диагностике МПС-I практикующими педиатрами и детскими ревматологами, с последующим их внедрением в клинические рекомендации и для определения значимости рассмотренных критериев был проведен медико-статистический анализ данных, полученных путем анкетирования, который включил следующие методы:
- Описательная статистика:
- частотные характеристики рассматриваемых критериев – простейший метод анализа категориальных переменных. Цель метода – оценить, каким образом различные группы наблюдений распределены в выборке или как распределено значение признака на интервале от минимального до максимального значения;
- средние значения объединенных показателей. Некоторые признаки были объединены по качественной характеристике и функциональной связи признаков (табл. 1).
- Дисперсионный анализ, направленный на поиск зависимостей в данных, – исследование связи между несколькими качественными переменными и одной зависимой количественной.
- χ2-критерий Пирсона – для проверки гипотезы о различии в частоте встречаемости признака у пациентов с МПС-I и у пациентов с неподтвержденным диагнозом. Этот критерий актуален в нашей выборке при гипотезе «между двумя переменными нет зависимости». Таким образом, метод позволил оценить статистическую значимость различий нескольких относительных показателей. Анализ был проведен на выборке из 16 пациентов с МПС-I и из 59 детей с подозрением на генетический диагноз.
- Критерий «отношение шансов», который позволил определить, насколько отсутствие или наличие определенного исхода связано с присутствием или отсутствием определенного фактора в конкретной статистической группе. Определенным фактором в данном анализе выступали признаки проявления МПС-I, а исходом – влияние этих признаков на возможное диагностирование наследственной патологии у пациентов. Чем выше этот показатель, тем теснее связь признака с заболеванием. Ранжированный список рассматриваемых признаков заболевания составлен по критерию «отношение шансов» по двум выборкам: по выборке из 16 пациентов, которым поставлен диагноз МПС-I, и по выборке из 59 пациентов с неподтвержденным диагнозом МПС.
- Кластерный анализ – градация множества исследуемых факторов и признаков на однородные в некотором смысле группы (кластеры). Он дал возможность произвести разделение признаков не по одному, а по ряду рассматриваемых критериев. Каждый признак принадлежит только одной группе.
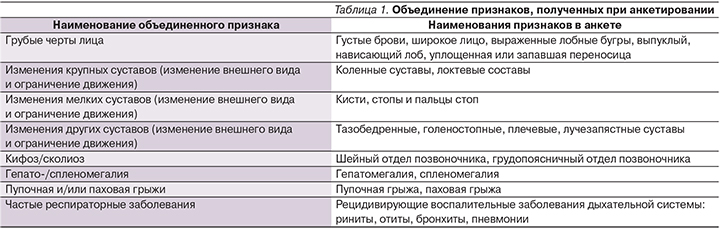
В табл. 1 приведены объединенные клинические признаки, характерные для больных МПС.
Результаты
Частота выявления МПС-I
Для создания математической модели были использованы данные исследования протестированных 738 пациентов с подозрением на МПС, среди которых у 7 пациентов подтвердили МПС-I. Можно заметить, что 1 пациент с МПС-I приходится на 105 пациентов в выборке (в группе). Следовательно, частота выявления МПС-I среди пациентов с подозрением на МПС составила 10:1050. Принимая во внимание данную частоту, становится ясно, что вероятность выявления пациентов с МПС в нашей выборке из 59 пациентов довольно мала. Это объясняет тот факт, что пациентов с МПС-I в выборке из 59 человек обнаружено не было.
Выявление критериев, помогающих заподозрить у пациентов МПС-I
Для того чтобы определить, какие признаки в целом характерны для МПС-I, была вычислена частота встречаемости каждого отдельного признака среди 16 пациентов с МПС-I (табл. 2).
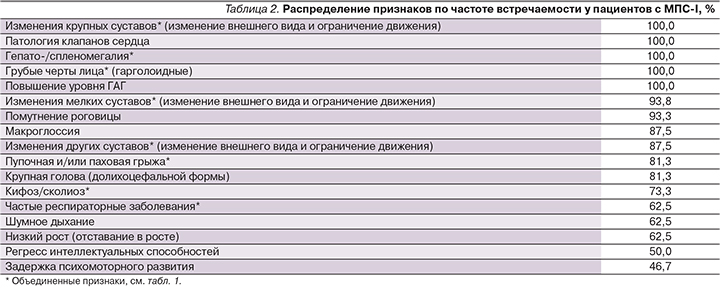
Стоит отметить, что в группу «крупные суставы» попали только локтевые и коленные суставы. Такой выбор основан на факте, согласно которому эти суставы были изменены (контрактуры) у всех 16 пациентов в возрасте до года. Ограничения движения в суставах возникают в среднем у пациентов с синдромом Гурлер в 10 месяцев [16], а т.к. первыми страдают локтевые и коленные суставы, было вполне разумно разделить признаки поражения крупных суставов на две группы: поражение локтевых вместе с коленными суставами и всех остальных (см. табл. 1).
При синдроме Гурлер гепатомегалия и спленомегалия, нарушение со стороны клапанов сердца, наличие контрактур, пупочной или паховой грыжи и помутнение роговицы были выявлены у детей в возрасте до года [16, 17]. Поскольку средний возраст исследуемых пациентов с МПС-I составил чуть больше 8 лет, вполне очевидно, что кроме пупочной/пааховой грыжи и помутнения роговицы (81,3 и 93,3% соответственно) остальные признаки из вышеперечисленных проявились у 100% наших пациентов (см. табл. 1).
Грубые черты лица также были отмечены у 100% пациентов с МПС-I, что согласуется с выводами других исследований [6]. Некоторые признаки были объединены в одну группу (например, поражения локтевых и коленных суставов объединены в признак «поражение крупных суставов»).
Для сравнения также была подсчитана частота встречаемости тех же признаков у пациентов с подозрением на МПС, но не подтвержденным диагнозом (59 пациентов с МПС-I) (табл. 3). Из приведенных данных видно, что есть признаки, которые часто встречаются у пациентов с МПС и в то же время достаточно редко проявляются у пациентов без этого диагноза. Например, помутнение роговицы и изменения со стороны клапанов сердца – самые редко встречающиеся признаки у 59 пациентов с подозрением на МПС (3,9 и 14,3% соответственно). Макрогласия, гепато/спленомегалия и пупочная и/или паховая грыжи проявлялись тоже редко и были выявлены у 18,5%, 18,8 и 25% пациентов соответственно (табл. 3), в то время как эти признаки были обнаружены более чем у 81% пациентов с подтвержденным МПС-I (см. табл. 2).
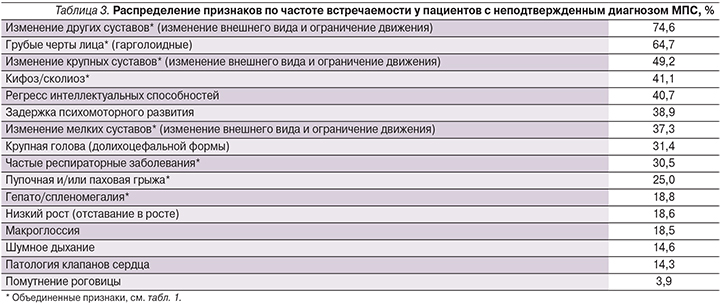
Довольно высокая частота встречаемости обнаружена для следующих признаков: изменение других суставов и изменение крупных суставов. Это объясняется следующим: все 59 пациентов из выборки имели ревматоидные симптомы без воспалительных процессов. Таким образом, они изначально обладали широким спектром нарушений опорно-двигательного аппарата.
В литературе достаточно часто одним из первых симптомов при МПС-I упоминаются респираторные заболевания верхних дыхательных путей, такие как отиты и риниты [5, 6, 8, 9]. В нашем исследовании мы собирали данные о наличии каких-либо частых респираторных заболеваний, не рассматривая их по отдельности. Однако именно частые рецидивирующие отиты могут служить сигналом о наличии генетического заболевания у ребенка. Таким образом, в названии критерия для выявления МПС-I у детей должны фигурировать частые отиты как симптом, на который стоит обратить особое внимание.
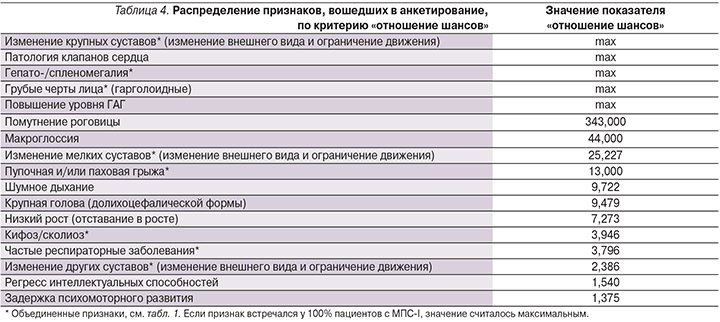
Следующим этапом был проведен анализ показателя «отношение шансов» для каждого из исследуемых признаков в двух выборках пациентов с подтвержденным диагнозом «мукополисахаридоз» и без выявленного МПС (табл. 4). Значение показателя «отношение шансов» тем выше, чем чаще встречается рассматриваемый признак у пациентов с МПС-I и реже проявляется у пациентов с неподтвержденным диагнозом. В тех случаях, когда признак проявлялся у 100% больных МПС-I (см. табл. 2), значение показателя «отношение шансов» считалось максимальным и эти признаки были автоматически перенесены в верхнюю часть таблицы (табл. 4).
Обсуждение
Очевидно, что признаки со значением «max» имеют наибольшую значимость для выявления заболевания (см. табл. 4). Повышенный уровень ГАГ стоит рассматривать обособленно, т.к. в 90% случаев наличие этого признака будет говорить о выявлении какого-либо из МПС (кроме IX) [18]. Однако этого признака недостаточно, т.к. уровень ГАГ также повышен при сахарном диабете 2 типа, артрите и муколипидозе [18]. Признак «помутнение роговицы» набрал самый высокий балл «отношение шансов» (кроме «max»), что, несомненно, говорит о сильной связи этого признака с МПС-I.
Другими словами, обращая свое внимание в первую очередь на помутнение роговицы у детей до года, педиатры сумеют распознать МПС-I на ранних стадиях развития [16]. Крупные и мелкие суставы сильнее подвержены изменениям при «мягких» типах болезни (синдром Шейе и Гурлер–Шейе), и указанные нарушения суставов являются в 54% случаев первым симптомом и причиной для обращения к врачу [14]. Таким образом, изменения в крупных («Крупные суставы» см. табл. 1) и мелких суставах должны входить в критерии распознавания МПС-I. Для больных синдромом Гурлер в 42% случаев первым толчком для обращения к врачу послужило шумное дыхание [14]. То есть признаку «шумное дыхание» стоит уделить дополнительное внимание в случае подозрения на МПС-I, несмотря на то что этот признак набрал не самый высокий балл «отношение шансов» в нашем исследовании (табл. 4).
Cистема присвоения баллов критериям
Диагностика МПС затруднена тем фактом, что заболевание не всегда проявляется явными признаками в дебюте [14]. Идентификация болезни в раннем возрасте затруднена, особенно когда педиатры, неонатологи, хирурги, ортопеды и другие специалисты не связывают различные признаки и симптомы между собой и рассматривают их как отдельные проявления [14, 16, 17]. В конечном итоге диагноз МПС не ставят и пациенту проводят симптоматическое лечение без эффекта.
Для усовершенствования и самое главное – для ускорения диагностики заболевания МПС-I у пациентов необходимо сформировать группы критериев с указанием значимости каждого из них, что поможет с высокой вероятностью поставить диагноз. К этим критериям относятся признаки, выделенные нами ранее и определенные на основе оценки показателя «отношение шансов» (см. табл. 4). Весь список критериев можно разбить на три группы в зависимости от срока появления этих симптомов и критерия «отношение шансов». Так как некоторые признаки проявляются в более раннем возрасте (паховые грыжи проявляются в среднем в возрасте 2 месяцев [16]), вполне очевидно, что такие признаки окажутся более важными для распознавания заболевания до начала его прогрессирования. В итоге в I группу попали признаки, самые важные для выявления МПС-I, каждому из которых присвоена значимость в 3 балла. Во II группу отнесли менее важные признаки, и их значимость равна двум баллам, в III группу – самые, на наш взгляд, не значимые для выявления МПС-I признаки, равные 1 баллу каждый (табл. 5).
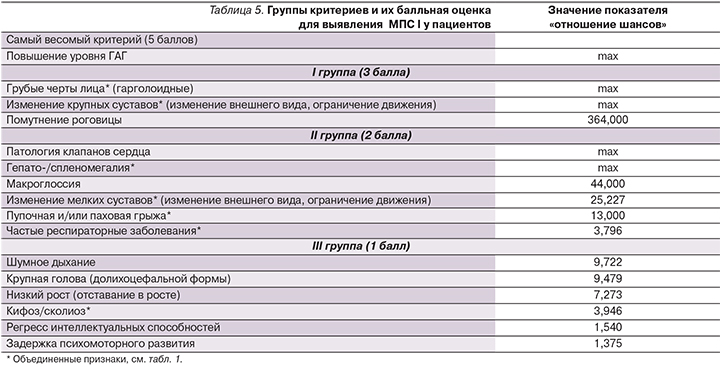
Исходя из вышеуказанной балльной системы, врач, осматривая пациента, может проставить баллы за наличие каких-либо признаков или симптомов. После чего, просуммировав эти баллы, он может оценить результат по следующей методике: если набранное число баллов больше или равно 15, врач принимает решение о направлении этого пациента на анализ ГАГ (если он не проводился ранее), энзимодиагностику и затем на молекулярно-генетическое тестирование.
Для валидации данной системы критериев был проведен анализ признаков у 59 пациентов с неподтвержденным диагнозом МПС-I. Он показал, что число баллов, набранных пациентами в данной выборке, редко достигало порогового значения (15 баллов) и только у 4 (6,7%) пациентов результат был не менее 15 баллов. Это говорит о том, что использование данного подхода может способствовать снижению процента пациентов с ложноположительным диагнозом. С другой стороны, система критериев была апробирована на 23 пациентах с подтвержденным диагнозом МПС-I. Подсчет набранных баллов показал, что практически все пациенты с МПС-I набрали более 15 баллов и лишь только 2 пациента с синдромом Гурлер–Шейе набрали по 15 баллов. Известно, что при синдроме Гурлер–Шейе и Шейе изменения органов могут появляться значительно позже [8], поэтому и число набранных баллов по нашей системе критериев при «мягких» типах МПС-I типа будет меньше, чем при синдроме Гурлер.
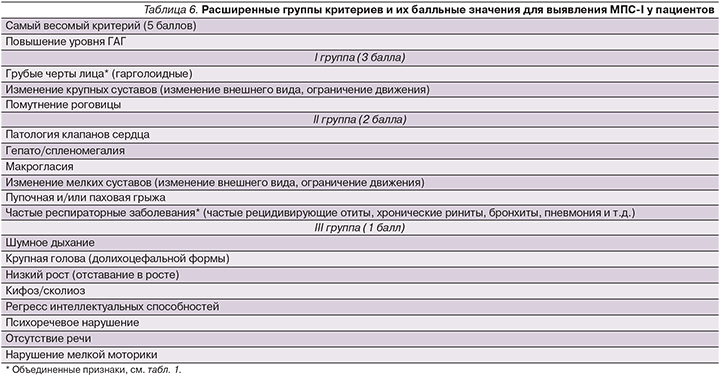
В результате исследования удалось сгруппировать наиболее важные признаки, позволяющие заподозрить МПС-I у конкретного пациента. Внедрив балльную систему для каждой группы признаков и симптомов, можно объективно оценить вероятность диагностирования МПС-I у пациентов в раннем возрасте. Наиболее характерные признаки относятся к первой группе, далее по нисходящей идет вторая группа, потом третья (табл. 6).
Заключение
В настоящей работе был проведен анализ данных трех групп пациентов: с подозрением на МПС-I, с подтвержденным диагнозом МПС-I и пациентов скрининговой программы диагностики МПС в РФ. Результаты исследования продемонстрировали, что частота выявления пациентов с МПС-I среди лиц с подозрением на МПС при генетическом анализе равна 1:105; соответственно частота выявления МПС-I среди таких лиц составляет 10:1050. Факт невыявления ни одного пациента с МПС из группы, включившей 59 пациентов с подозрением на МПС, подтверждает полученную частоту выявления таких пациентов. Суммируя баллы за каждый выявленный признак или симптом, можно с определенной долей вероятности предположить заболевание МПС-I у конкретного пациента. Однако поставить окончательный диагноз возможно, лишь опираясь на клинические проявления и результаты лабораторных анализов (определение активности фермента альфа-L-идуронидазы и генетическое обследование).
В заключение можно утверждать, что полученные группы критериев, представленные в табл. 6, могут способствовать улучшению диагностики пациентов с МПС-I.
