Мукополисахаридозы (МПС) представляют собой группу редких генетических заболеваний, которые развиваются вследствие недостаточности лизосомных ферментов обмена гликозаминогликанов, что приводит к их отложению в соединительной ткани и развитию полиорганной патологии. Клиническая картина заболевания характеризуется нарушением проходимости верхних дыхательных путей, хроническими инфекционновоспалительными заболеваниями носоглотки, пневмониями, клапанными пороками, поражением коронарных и системных артерий, миокардиопатией, артериальной гипертензией, гепатоспленомегалией, множественными дизостозами, патологией суставов, нарушением роста, сдавлением спинного мозга, компрессионными невропатиями, гидроцефалией, умственной отсталостью, апноэ во сне, помутнением роговицы, ретинопатией, глаукомой, снижением слуха и другими нарушениями [1–5]. Поскольку в прогрессирующий патологический процесс вовлекаются все органы и системы организма, МПС являются объектом полидисциплинарного медицинского подхода.
До начала ХХI в. дети с МПС получали лишь симптоматическое лечение, которое существенно не влияло на прогрессирующий характер заболеваний, а продолжительность их жизни не превышала 30 лет [6, 7]. Благодаря успехам генной инженерии и фарминдустрии, позволившим приступить к синтезу ферментных препаратов в промышленных масштабах, с 2006 г. началось активное патогенетическое лечение отдельных форм МПС (I, II и VI типов) с помощью регулярных инфузий ферментозамещающих (ФЗ) препаратов. Многоцентровые рандомизированные исследования показали, что ферментозаместительная терапия достоверно улучшает объем пассивных и активных движений, уменьшает объемы паренхиматозных органов, улучшает проходимость верхних дыхательных путей, что препятствует дальнейшему прогрессированию заболевания [8–13]. Таким образом, за последние годы произошел кардинальный переворот в отношении некоторых форм МПС: новые виды лечения и пересмотр прогноза заболевания в позитивную сторону полностью изменили медицинскую стратегию и тактику ведения этих пациентов.
В данной статье мы обобщаем опыт внедрения ферментозаместительной терапии (ФЗТ) в нашем учреждении и представляем модель организации многоэтапной комплексной помощи детям с МПС.
Первые пациенты с МПС поступили в центр для проведения инфузий ферментных препаратов в 2008 г. На настоящий момент НЦЗД РАМН обладает наибольшим опытом ФЗТ детей с МПС в России. В течение 4 лет под нашим наблюдением находились 60 пациентов с МПС (табл. 1).

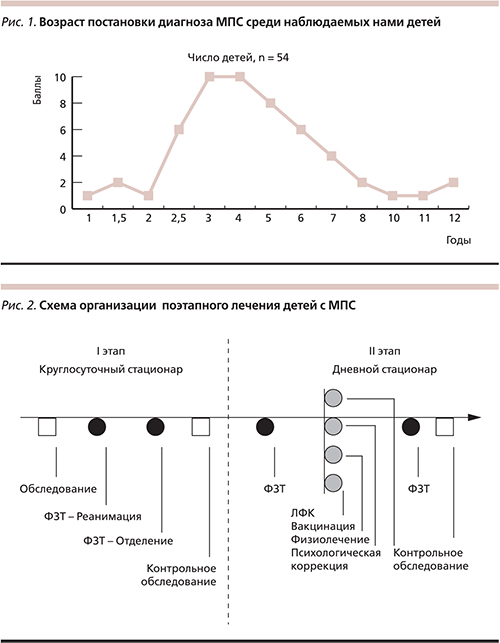
Эффективность ферментотерапии в предотвращении развития многочисленных органических поражений тем выше, чем раньше начато лечение; соответственно, перед педиатрами стоит задача как можно раньше выявить наличие МПС у ребенка [14–18]. В европейских странах и США, как правило, к ФЗТ приступают уже на первом, реже – втором, году жизни ребенка. К сожалению, в России диагноз МПС выставляется очень поздно. По нашим данным, в России до 2 лет МПС был диагностирован всего в 7 % случаев (т. е. у 4 из 54 детей), а в среднем диагноз выставлялся в 4,74 года (рис. 1). Таким образом, чаще всего ферментотерапия начиналась поздно – к моменту, когда патологические изменения приобретали устойчивый и взаимоусугубляющий характер. По этой причине четыре случая завершились летальным исходом, что было обусловлено крайне тяжелым состоянием детей ввиду длительного стажа заболевания к моменту обращения к специалистам НЦЗД РАМН, даже несмотря на начало ФЗТ в круглосуточном стационаре одному из них. В связи с этим имеется острая необходимость информационной работы с широким кругом педиатров, включая специалистов узкой специализации: с 2008 г. по настоящее время под нашим наблюдением находились 60 пациентов с различными типами МПС, однако не все формы МПС доступны ФЗТ.
Пациенты с МПС I, II, и VI типов получают ФЗТ, а дети с МПС III и IV типов – наблюдение и симптоматическое лечение.
Специалисты научно-клинической группы по лечению МПС разработали и реализовали алгоритм лечения больных МПС I, II, и VI типов начиная с первых инфузий ферментозаместительными препаратами, суть которого заключается в поэтапном лечении и наблюдении пациентов (рис. 2). Каждый этап базируется в определенном структурном подразделении НЦЗД РАМН, эти структурные подразделения поддерживают связь в рамках единой научно-клинической группы и передают друг другу пациента в зависимости от клинической необходимости.
Первый этап наблюдения проводится в психоневрологическом или кардиологическом отделениях круглосуточного стационара в зависимости от ведущего профиля заболевания. В этих отделениях есть возможность круглосуточного наблюдения за ребенком после первых инфузий ферментного препарата и оказания квалифицированной медицинской помощи вплоть до срочного перевода в реанимационное отделение в случае возникновения острых аллергических реакций (крапивницы, отека Квинке, анафилактического шока), колебаниях артериального давления, нарушении ритма сердца, частоты дыхания, гипертермии, артралгиях, болях в животе, диспепсических расстройствах, боли за грудиной и др.
Суть первого этапа заключается в комплексном обследовании и инициации ФЗТ конкретного ребенка, отработке технологии первых инфузий ФЗ препаратов с достижением стабильного состояния и отсутствия каких-либо жизнеугрожающих или других серьезных осложнений у пациента, характерных для первых инфузий белковых препаратов.
Вначале проводится полное исходное обследование дыхательной, сердечнососудистой и нервной систем, когнитивной сферы, желудочно-кишечного тракта, органов зрения, ЛОР-органов, опорно-двигательного аппарата. Кроме того, оценивается аллергический статус, что необходимо для адекватной подготовки пациента к первым инфузиям (проведение премедикации антигистаминными препаратами, своевременное оказание помощи в случае развития острой аллергической реакции).
По окончании обследования начинается еженедельное внутривенное введение ФЗ-препарата с постепенным увеличением скорости введения лекарственного вещества по стандартной схеме. Первые две инфузии осуществляются в условиях реанимационного отделения в связи с возможностью развития таких жизнеугрожающих ситуаций, как анафилактический шок, отек Квинке, нарушения ритма сердца и пр. В отсутствие побочных реакций третья и четвертая инфузии уже проводятся в условиях клинического отделения. Особое внимание необходимо обратить на состояние ребенка во время проведения шестой и седьмой инфузий, поскольку, по нашим наблюдениям, именно в этот период наиболее высока вероятность развития реакций непереносимости ФЗТ.
Во время госпитализации в круглосуточном стационаре проводится обследование пациента с целью фиксации краткосрочных эффектов ФЗТ. За прошедший период на первом этапе ФЗТ впервые получил 31 ребенок (29 – в отделении психоневрологии, 1 – в отделении хирургии и 1 в отделении кардиологии), проживающий в различных регионах страны. В психоневрологическое отделение круглосуточного стационара для проведения плановых контрольных обследований также госпитализируются дети с МПС, проживающие вне Московского региона, как получающие ФЗТ по месту жительства, так и не получающие ее ввиду недостаточного финансирования.
После завершения первого этапа ребенок направляется на второй этап лечения в Реабилитационный центр НЦЗД РАМН, если он проживает в Московском регионе. Жители других регионов направляются на дальнейшее лечение по месту проживания с обязательным посещением НЦЗД РАМН 1 раз в 3 месяца для контрольного обследования.
На втором этапе пациент госпитализируется в кардиологическое отделение реабилитационного центра. Суть второго этапа заключается в пролонгированном комплексном восстановительном лечении, включающем ФЗТ, которое осуществляется в режиме дневного стационара, когда ребенок посещает центр исключительно для проведения лечебно-реабилитационных процедур. Для длительно наблюдающихся пациентов форма дневного стационара существенно более удобна по сравнению с круглосуточным стационаром, поскольку исключаются реакции госпитализма и внутрибольничные инфекции, экономится время семьи и ребенка. На втором этапе контроль всего процесса лечения и наблюдения ребенка с МПС осуществляется врачом-куратором, который применяет комплексный подход, аналогичный для любой другой хронической полиорганной патологии.
Дети, проживающие недалеко от реабилитационного центра, получают инфузии и другие виды помощи исключительно в нашем реабилитационном центре неопределенно долго – большинство пациентов продолжают получать регулярные инфузии и по настоящее время.
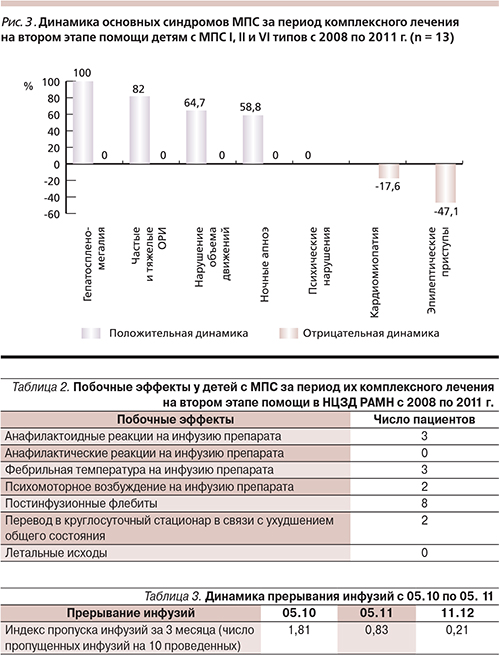
В условиях дневного стационара пациенты получают не только ФЗТ, но и другие методы лечения: лечебную физкультуру (ЛФК), физиотерапию, психолого-педагогическую коррекцию и занятия с логопедом. ЛФК и физиотерапия позволяют закреплять и развивать положительные изменения состояния опорнодвигательного аппарата, таким образом повышая эффективность лечения. Физиотерапевтические процедуры эффективны также в отношении хронических инфекционновоспалительных заболеваний верхних дыхательных путей. Занятия по психолого-педагогической коррекции направлены на социально-бытовую адаптацию детей и работу с семьей. В центре вакцинопрофилактики проводится вакцинация детей. В настоящее время детям с МПС как группе высокого риска по инфекциям помимо основного календарного плана начато проведение дополнительной вакцинации (против пневмококковой и гемофильной инфекций).
Помимо этого проводится регулярное контрольное обследование пациентов. Для контрольного обследования и вакцинопрофилактики используется база консультативнодиагностического центра, тесно интегрированного с реабилитационным центром. В процедуру контрольного обследования помимо комплекса биохимических анализов включены исследование функции внешнего дыхания, магнитно-резонансная томография МРТ головного и спинного мозга, электороэнцефалография, полисомнография, акустические и зрительные вызванные потенциалы, исследование слуха и др. При необходимости проводятся консультации любого специалиста узкого профиля. Для мониторинга уровня гликозаминогликанов в моче, определения активности лизосомной идуронатсульфатазы в лейкоцитах или в культуре фибробластов, ДНКдиагностики образцы биологических жидкостей всех пациентов направляются в Национальный медикогенетический центр.
Особо следует подчеркнуть, что весь комплекс лечения и обследования пациента проводится на одной территории по плану врача-куратора в режиме дневного стационара. Однако в случае развития негативных сценариев течения заболевания или отдаленных жизнеугрожающих осложнений есть возможность оперативного перевода ребенка в отделения круглосуточного стационара или в реанимационное отделение.
За 4 года работы научноклинической группы лечение с ФЗТ на первом этапе получил 31 пациент, а на втором – 17. За это время на втором этапе лечения в режиме дневного стационара было проведено более 900 инфузий, 200 сеансов массажа и занятий лечебной физкультуры, 600 физиопроцедур, 300 занятий с психологами, логопедами и дефектологами.
В результате у наблюдаемых пациентов на втором этапе была достигнута стойкая положительная динамика. Полученные нами данные по динамике течения основных клинических синдромов совпадают с представленными в зарубежной литературе эффектами ФЗТ для поздно диагностированных случаев: физическое самочувствие пациентов значительно улучшается в основном за счет снижения частоты острых респираторных инфекций (ОРИ), улучшения проходимости верхних дыхательных путей, снижения частоты ночных апноэ, уменьшения гепатоспленомегалии и повышения объема двигательной активности (рис. 3) [8, 11, 13]. Снижение частоты ОРИ, облегчение их течения, а также наличие возможности экстренно госпитализировать ребенка в круглосуточный стационар для интенсивной терапии осложнений ОРИ позволили избежать летальных исходов на протяжении всего периода комплексного лечения пациентов на втором этапе.
Всего трижды более чем за 900 инфузий ФЗ-препаратов отмечены анафилактоидные реакции, причем в одном случае – повторные, купированные без прерывания инфузий. Более тяжелых аллергических осложнений отмечено не было. Какие-либо осложнения зарегистрированы всего у 47 % пациентов, чаще имели место постинфузионные флебиты, своевременно купированные с помощью местной терапии; другие реакции отмечены единично или кратковременной серией и были также купированы (табл. 2).
Эффективная профилактика инфекционно-воспалительных и аллергических обострений, постинфузионных флебитов, а также тактика разумного ограничения и оптимизации списка противопоказаний к инфузиям позволили за год минимизировать пропуски ферментозаместительных инфузий более чем в 3 раза – в настоящее время пропуски инфузий происходят около 2 раз на 100 проведенных инфузий (табл. 3).
Таким образом, в течение 4 лет специалисты научно-клинической группы по МПС успешно провели введение ФЗ-препаратов 31 ребенку с различными формами МПС, а также разработали и апробировали оригинальный алгоритм контрольного обследования и комплексного лечения детей с МПС в режиме дневного стационара. С целью актуализации проблемы, а также повышения информированности педиатров в вопросах диагностики и лечения МПС руководство центра регулярно организует научно-практические конференции, симпозиумы, на которых выступают с докладами представители научно-клинической группы по МПС НЦЗД РАМН и других российских учреждений. Кроме того, его специалисты неоднократно выезжали с информационно-обучающими программами в медицинские учреждения различных российских регионов, где проводили обучение технологии ФЗТ, мастер-классы по диагностике, тактике ведения и лечению детей с МПС. Большое внимание уделяется работе с пациентской организацией: проводятся совместные акции (например, ежегодные праздничные акции для детей с МПС, приуроченные к международному дню защиты детей), организуются совместные конференции, доклады и выступления. Анализируя работу научно-клинической группы, можно констатировать, что она занимается различными видами деятельности и с учетом количества наблюдаемых пациентов фактически выполняет роль ведущего центра лечения и наблюдения детей с МПС в России.
Подводя итоги, можно констатировать, что, несмотря на некоторые сохраняющиеся системные организационные трудности, научно-клинической группе по МПС удалось успешно внедрить ферментотерапию детей с МПС в России и наполнить новым содержанием всю технологию наблюдения и лечения детей с МПС, предоставив им возможности комплексной реабилитации по всем направлениям их развития.
В настоящее время вводятся и планируются к вводу новые дополнительные технологии лечения детей с МПС, к числу которых относятся внедрение в практику порт-систем и налаживание паллиативной помощи. Паллиативная помощь, адресованная детям с любыми формами МПС, – это подход, реализуемый на разных уровнях медицинской и социальной помощи, целью которого является улучшение качества жизни больных и членов их семей, оказавшихся перед лицом угрожающего жизни заболевания. Эта цель достигается путем предупреждения и облегчения страданий благодаря раннему выявлению, тщательной оценке, купированию боли и других тягостных физических симптомов, а также оказанию психосоциальной и духовной поддержки. К паллиативным мероприятиям относят адекватное обезболивание и купирование других тягостных симптомов, психологическую поддержку больного и ухаживающих за ним родственников, выработку отношения к смерти как к закономерному этапу пути человека, удовлетворение духовных потребностей больного и его близких, решение социальных, юридических и этических вопросов, которые возникают в связи с тяжелой болезнью и приближением смерти человека.
Установка порт-систем для инфузий позволяет избегать еженедельных катетеризаций вен, что облегчает технологию инфузий, избавляет от образования флебитов и экономит медицинские силы.
Помимо этого в настоящее время по завершении этапа клинической организации помощи детям с МПС мы приступили к созданию реестра пациентов с МПС в России и его унификации с международным реестром. Научно-клиническая группа продолжает совершенствовать и наполнять новыми технологиями комплексную помощь детям с МПС, используя последние достижения всех областей медицины.
