
Введение
Боковой амиотрофический склероз (БАС) - неуклонно прогрессирующее и смертельное нейродегенеративное заболевание, которое сопровождается поражением центральных и периферических мотонейронов, двигательным дефицитом, нарушениями речи, глотания, алиментарной и дыхательной недостаточностью.
Впервые генетическая природа БАС была доказана в 1993 г. С тех пор открыты мутации в гене суперок-сиддисмутазы-1 (SOD1), липосаркомы (FUS/TLS), TDP-43/TARDBP, C9orf72, ангиогенина (ANG), везикуло-ассоциированного белка В (VAPB), FIG4, оптиневрина, валозин-содержащего белка, убиквилина-2, профилина-1, др. [1-8].
В Российской Федерации генетика БАС впервые изучалась в 1999-2003 гг. В.И. Скворцовой, Г.Н. Левицким и соавт. Были обнаружены мутации в гене SOD1 у больной крайне медленно прогрессирующим поясничным дебютом БАС в классическом варианте (гомозиготная D90A), а также у больной типичным быстрым прогрессированием поясничного дебюта БАС в классическом варианте (гетерозиготная D90A). Мутации D90A впервые были открыты в Швеции P.M. Andersen [9]. У обеих российских больных обнаружен т.н. скандинавский гаплотип, что привело к суждению о наличии другого неизвестного каузативного генетического или средового фактора БАС у второй пациентки [10, 11].
Также была описана пациентка с мутацией G12R с быстропрогрессирующим поясничным дебютом БАС в пирамидном варианте, при этом более пожилой возраст больной (62 года) и быстрый темп прогрессирования отличали ее от выборки итальянских пациентов, у которых заболевание начиналось в четвертой декаде жизни и медленно прогрессировало [11-13].
Сотрудниками Научного центра неврологии РАН были обследованы 285 российских пациентов с БАС, включая 260 пациентов со спорадической и 25 с семейной формами, на предмет носительства мутаций в генах SOD1, C9orf72, TARDBP, ANG и др., а также на наличие ассоциаций с полиморфными сайтами в генах ATXN2 ^uCAG) и VEGF (-2578С/А). Молекулярно-генетический анализ выполняли с использованием методов прямого секвенирования, фрагментного анализа и полимеразной цепной реакции (ПЦР) в режиме реального времени. На последнем этапе оценивали редкие кандидатные гены БАС с использованием секвенирующей панели нового поколения (NGS — next generation sequencing). Суммарная частота выявленных мутаций в обследованной когорте пациентов с БАС составила 9,5%. Наиболее частыми оказались повреждения в генах SOD1 (24,0% при семейной форме БАС и 4,6% при спорадической) и C9orf72 (патологическая экспансия гексануклеотидных повторов в нем обнаружена в 1,8% случаев БАС, все случаи спорадические). Мутаций в гене TARDBP не обнаружено. Мутации в гене ANG выявлены у 1,05% обследованных больных БАС (все случаи спорадические) [14].
В 2011 г. высокая экспансия гексануклеотидных повторов GGGGCC (G4C2) в некодирующей области 9-й хромосомы с открытой рамкой считывания 72(C9ORF72) была идентифицирована как самая частая генетическая мутация у пациентов с БАС-лобно-височной деменцией. Ген C9orf72 состоит из 12 экзонов, из них 2 некодирующих (1a и 1b). Размер G4C2 экспансии у больных может достигать 600-2000 повторов [3].
Общими механизмами нейронной дисфункции при БАС и лобно-височной деменции считаются дефицит биогенеза рибосомной РНК (рРНК) и утрата целостности ядрышка - состояния, известного как ядрышковый стресс, которое является общей чертой нейродегенеративных заболеваний. Мутация C9orf72 при лобно-височной деменции препятствует функционированию ядрышка путем транскрипции и дипептидных повторов, продуцируемых гексануклеотидной экспансией.
Мутантная РНК и белки, полученные при генетических мутациях, могут влиять на различные стадии биогенеза рРНК. С учетом этих особенностей ядрышковый стресс все чаще оценивается как потенциальный механизм, поддерживающий или лежащий в основе нейродегенерации. Все большее число исследований подтверждает наличие ядрышкового стресса при C9orf72 HRE-вызванных нарушениях у пациентов в моделях БАС-лобно-височной деменции.
В европейской и североамериканской популяциях самым частым повреждением является экспансия гексануклеотидных повторов GGGGCC в интроне 1 гена C9orf72, она выявляется при семейной форме БАС в среднем в 40% случаев, при спорадической - в 7-11%. После SOD1 ген C9orf72 - наиболее частый по повреждению в российской выборке больных БАС. В российской популяции частота встречаемости мутаций в гене C9orf72 составляет 15,0% при семейной форме и 2,5% при спорадической. В ходе одного из исследований в российской популяции у 5 пациентов (2 мужчин и 3 женщин) со спорадической формой БАС выявлена гетерозиготная мутация по типу гексануклеотидной GGGGCC-экспансии в интроне 1 гена C9orf72. Число повторов в гене у всех исследуемых превышало патологический порог в 50 копий. Возраст дебюта болезни у данных пациентов варьировался от 38 до 65 лет и не имел доказанной связи с числом копий GGGGCC-повторов, у всех больных БАС быстро прогрессировал и переходил в генерализованную форму к концу первого года болезни. Частота мутаций гена C9orf72 у пациентов с семейной формой БАС высока в Бельгии и Греции - до 50%, в Финляндии - 46,4% [15, 16].
В исследовании, проанализировавшем результаты генетического тестирования 254 больных БАС, проживающих в Италии, за последние 20 лет показано, что экспансия C9orf72 дидезокси-методом (секвенированием по Сэнгеру) была выявлена у 5,5% больных - в 17,6% семейного БАС, 3,3% спорадических случаев и в 5,0% случаев с неизвестным семейным анамнезом. Возраст обследуемых составил 57,1±8,1 года [17].
В немецкой популяции 301 пациент с семейной формой БАС был обследован на предмет мутации в гене C9orf72: у 75 пациентов обнаружена экспансия гексануклеотидных повторов [18].
Цель настоящей работы: молекулярно-генетическое обследование больных БАС из российской популяции на предмет наличия мутаций в генах SOD1, ANG, TARDBP, VAPB, C9orf72.
Материал и методы
С 2013 по 2015 г. были обследованы 60 пациентов с БАС. Диагноз БАС ставился по результатам неврологического осмотра в соответствии с пересмотренными Эль-Эскориальскими критериями после проведения игольчатой и стимуляционной электромиографии (ЭМГ) и проведения магнитно-резонансной томографии (МРТ) отдела центральной нервной системы в проекции дебюта болезни и ростральнее [19]. У 10 больных имело место сочетание БАС и лобно-височной деменции, диагноз которой ставился согласно критериям D. Neary [20]. Дебют БАС диагностировался по классификации A.J. Hudson, вариант БАС - по классификации О.А. Хондкариана [21, 22]. Клиническое и нейрофизиологическое обследование проводилось на базе ООО «Клиника Глеба Левицкого», Москва.
Выделение геномной ДНК проводили с помощью набора AxyPrepTM Blood Genomic DNA Miniprep Kit 250-prep (AXYGEN) из образцов периферической крови пациентов, взятых в пробирки с этилендиаминтетрауксусной кислотой (ЭДТА).
Референсные последовательности исследованных генов были взяты из базы данных NCBI (http://www.ncbi. nlm.nih.gov/). Были выбраны праймеры для амплификации и последующего прямого автоматического секвенирования всех экзонов, включая области экзон-интронных соединений генов SOD1, ANG, VAPB, FUS, TARDBP. Специфичность и качество праймеров проверяли с помощью OligoCalc (http://biotools.nubic.northwestern.edu/ OligoCalc.html). Праймеры были синтезированы метоксифосфоамитидным методом в ЗАО «Евроген».
Амплификацию необходимых фрагментов геномной ДНК проводили методом ПЦР на программируемом термоциклере МС2 фирмы «ДНК-технология» (Россия) в 25 мл реакционной смеси следующего состава: 20-100 нг геномной ДНК или кДНК; по 0,25 мкМ каждого оригинального олигопраймера; по 200 мкМ каждого нуклеозидтрифосфата; 1,0 единица активности ДНК-полимеразы Biotaq («Силекс»); буфер для ПЦР (67 мкМ Tris-HCl; 16,6 мкМ (NH4)2SO4; 0,01% Twin-20; рН 8,8); 3,0-4,0 мкМ MgCl2; 20-30 мл минерального масла.
Последовательности использованных праймеров и условия проведения PCR представлены в табл. 1.
Амплификацию проводили в стандартных условиях при оптимальной температуре отжига. Для регистрации продуктов ПЦР использовали электрофорез в 3%-ном агарозном геле в присутствии бромистого этидия с последующей визуализацией с помощью документирующей системы GelDoc фирмы BIO-RAD в УФ-излучении.
Все фрагменты были секвенированы с обеих цепей ДНК с использованием Big Dye Terminator’s v. 1.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) и генетического анализатора ABI PRISM 3500xl (Applied Biosystems); анализ результатов секвенирования проводили с помощью программы Chromas. Анализ выявленных изменений нуклеотидной последовательности проводили с помощью Nucleotide BLAST (http:// blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_ TYPE=BlastSearch&BLAST_ SPEC=OGP_9606_9558&LINK_ LOC=blasthome#) и баз данных HGMD (http://www.hgmd.cf.ac.uk/ac/index. php), NCBI (http://www.ncbi.nlm.nih. gov/) и Ensembl (http://www.ensembl. org/index.html).
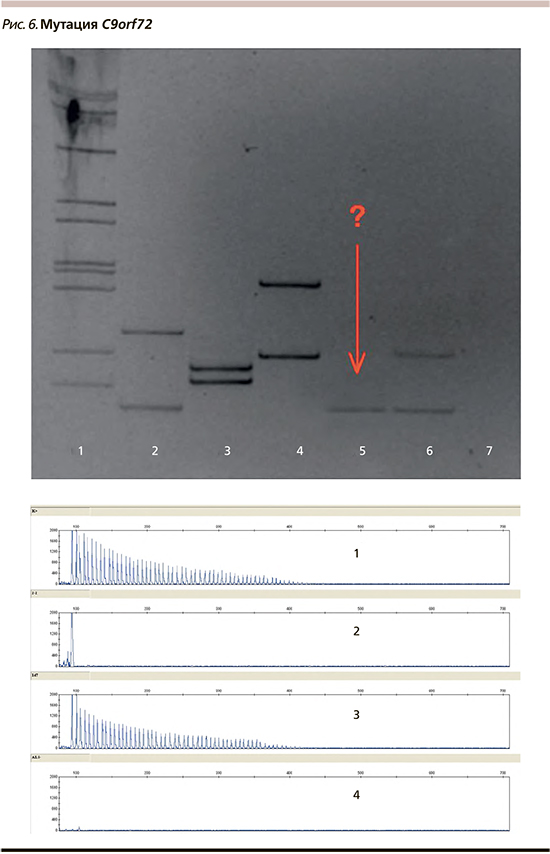
Для выявления увеличения числа GGGGCC-повторов в гене C9orf72 разработана двухэтапная система. На первом этапе осуществляется детекция коротких аллелей методом ПЦР. В случае выявления двух аллелей с нормальным количеством повторов ALS-TD1 исключается.
При выявлении одного короткого аллеля в геми-/гомозиготном состоянии проводится исследование методом ПЦР с праймером, комплементарным повтору, и праймером, меченным флуоресцентным красителем FAM, с последующим фрагментарным анализом на секвенаторе для подтверждения или исключения наличия длинного аллеля с увеличенным числом GGGGCC-повторов.
Последовательность праймеров представлена в табл. 2.
Амплификацию необходимых фрагментов ДНК проводили на программируемом плашечном термоциклере DNA Engine Tetrad 2 Cycler (Bio-Rad, USA) в 25 мл объеме реакционной смеси следующего состава: 0,1-1,0 мг геномной ДНК; 0,25 мкМ каждого оригинального олигопраймера; по 200 мкМ каждого нуклеозидтрифосфата; 1,0 единица MKM (NH4)2SO4; 0,01% Twin-20; pH 8,8); 1,6 М Betaine, 5% DMSO, 1 мкМ МgCl2. Температура отжига праймеров составила 58°С.
Разделение продукта реакции первого этапа проводили в полиакриламидном геле (соотношение АА/БА 29:1,8%) размером 20х20 см в камере для вертикального электрофореза «Helicon». Используемое напряжение: 270 В. Анализ осуществлялся на системе гель-документирования «BioRad».
Результаты амплификации второго этапа оценивали методом фрагментного анализа на приборе 3130 Genetic Analyzer (Applied Biosystems, Japan).
Результаты
Мутации в гене SOD1 выявлены у двоих больных БАС и у троих родственников первой из них. Мутации в гене ANG выявлены у одной больной БАС и у ее здорового сына. Мутация в гене VAPB выявлена у одного пациента с БАС. Мутация в гене TARDBP выявлена у одного пациента со спорадическим БАС и у двух с семейным БАС. Мутации гена C9orf72 выявлена у одной больной БАС с лобно-височной деменцией, а также у ее здоровой дочери.
Клинический случай 1
Больная Р. 43 лет больна в течение 3 лет. Проживает с семьей в Астраханской области. Заболевание началось со слабости в левой ноге. В неврологическом статусе нет когнитивных нарушений и расстройств черепных нервов, тетрапарез до плегии в стопах, парез до 2-3 баллов в передней группе мышц бедер, кистей с преобладанием в разгибателях, верхнем плечевом поясе с преобладанием слева, гиперрефлексия с рук с патологическими знаками, ахилловы рефлексы не вызываются, фасцикуляции в бедрах и плечах, атрофии мышц ног, кистей, верхнего плечевого пояса, чувствительность и координация, тазовые функции не нарушены. Диагноз: болезнь мотонейрона, БАС, поясничный дебют, классический вариант, быстрое прогрессирование.
Семейный анамнез. У больной была сестра, умершая от БАС в возрасте 43 лет, заболела в 39 лет. Имел место поясничный дебют БАС. У больной двое здоровых детей - 11 и 15 лет. У сестры больной остался здоровый сын 14 лет. Родителям больной 63 и 67 лет, они здоровы.
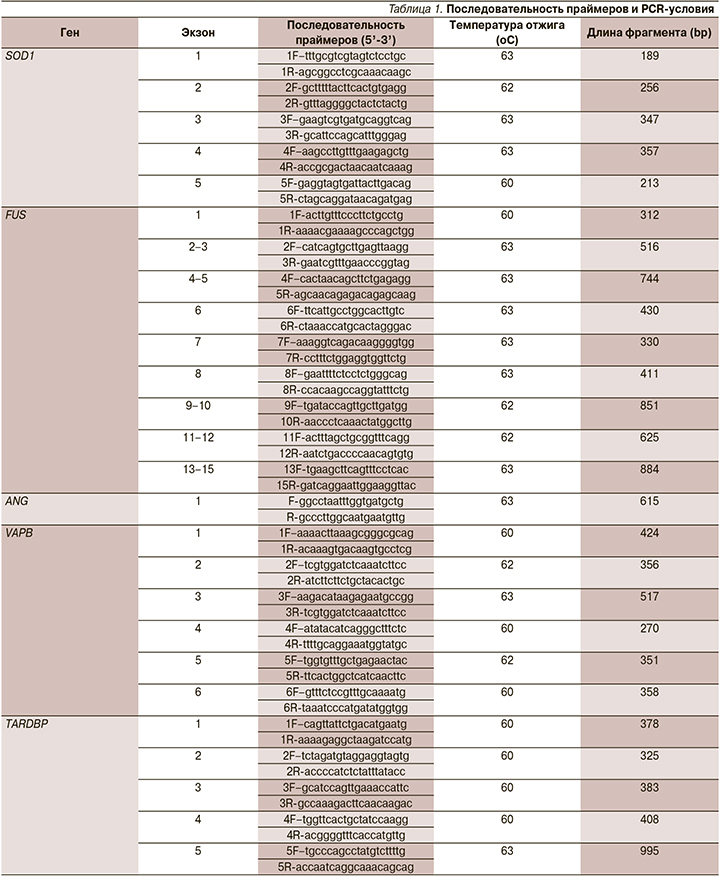
У больной (3) и обоих ее детей (1 и 2), а также у сына ее покойной сестры (6) выявлена мутация в гене SOD1 c.50G>C (p.17Gly>Ala) CM983777 NM_000454.4 в гетерозиготном состоянии (рис. 1, 2). У бабушки (4) и дедушки (5) больной, тети больной (7) данной мутации в гене SOD1 не выявлено. Родители больной обследованы не были.
Клинический случай 2
Больная Ц. 55 лет проживает в Вязьме Смоленской области, жалуется на слабость в руках и ногах, эпизоды замедления речи и редкие поперхивания, сложно дышать лежа на спине, генерализованные мышечные подергивания, легкие нарушения сна, часто плаксивость. Заболела в феврале 2014 г.: появилась слабость в левой кисти.
При игольчатой ЭМГ выявлены нейрональные изменения в мышцах рук и в меньшей степени - в ногах с признаками текущего денервационно-реиннервационного процесса. МРТ шейного отдела позвоночника - признаки остеохондроза, интрамедуллярных изменений нет. МРТ головного мозга - внутренняя гидроцефалия. Компьютерная томография (КТ) грудной клетки - объемное образование переднего средостения (целомическая киста перикарда? вилочковой железы?).
Семейный анамнез: мать больной умерла в возрасте 70 лет, были нарушения речи, поперхивания при глотании, трудности в самообслуживании, умерла после поперхивания. У больной дочь 33 лет и сын 28 лет, отец больной умер после инсульта в 62 года, у больной есть сестра 59 лет, второй сестре 60 лет, младшей - 53 года, у всех есть здоровые дети. Бабушка и дедушка по линии папы были долгожителями. Дедушка по линии мамы погиб на войне в возрасте примерно 40 лет, бабушка по линии мамы умерла в возрасте 64 лет, страдала нарушениями ходьбы, нарушением движений в руках и одышкой.
При осмотре в неврологическом статусе: когнитивные нарушения, расстройства черепных нервов нет, двусторонний рефлекс Маринеско, тетрапарез до глубокого пареза в руках, 3 балла в ягодицах, грубые атрофии мышц рук, единичные фасцикуляции в руках и ногах, гипорефлексия, координация и чувствительность не нарушены, одышка в положении лежа. Спирография - жизненная емкость легких (ЖЕЛ) 55%, форсированная жизненная емкость легких 51%, значительные рестриктивные нарушения.
ЭМГ - декремент-тест при разночастотной стимуляции при частоте 3 Гц, сила тока - 50 мА, первоначально получен декремент 16% от 1-го к 3-му ответу, далее декремент 44% на стимуляции частотой 50 Гц, далее декремент 33% на частоте 3 Гц, далее декремент 83% (т.е. получены феномены и посттетанического истощения, и посттетанической фасилитации).
Диагноз: семейная болезнь двигательного нейрона (боковой амиотрофический склероз, шейный дебют, сегментарно-ядерный вариант, быстрое прогрессирование - 21 балл в год по шкале ALSFRSR). Внутренняя атрофическая гидроцефалия. Сочетанная генерализованная форма миастении, вероятная тимома переднего средостения. Дыхательная недостаточность 2-й степени.
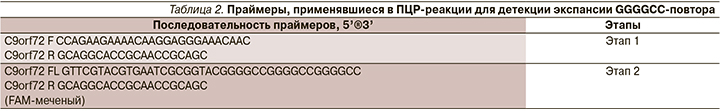
У больной выявлена мутация c.435G>C (p.145 Leu>Phe) CM 930687 NM_000454.4 в гене SOD1 в гетерозиготном состоянии. У детей пациентки данной мутации не выявлено (рис. 3).
Клинический случай 3
Больная А. 68 лет. Проживает в г. Каменско-Уральском Свердловской области. Жалуется на отсутствие речи, нарушение глотания жидкости и твердой пищи, снижение массы тела за время болезни (1,5 года) на 20 кг, слабость в руках и ногах, мышечные подергивания, слюнотечение, нарушение сна, плаксивость. В неврологическом статусе общемозговых и менингеальных симптомов нет, при оценке по Монреальской шкале когнитивных расстройств оценка 18 баллов (норма - 26-30), умеренные когнитивные нарушения. Жевание ослаблено, парез круговой мышцы рта, язык в полости рта с фасцикуляциями и атрофиями, слюнотечение, парез мягкого неба справа, оживлен мандибулярный рефлекс, рефлексы Маринеско с двух сторон, трипарез до 4 баллов в плечах и до 3 в разгибателях правой стопы, атрофии мышц стоп, кистей, вызванные фасцикуляции мышц верхнего плечевого пояса, сухожильные рефлексы с рук и коленные рефлексы оживлены, ахилловы снижены, подошвенные индифферентны, патологические кистевые рефлексы Якобсона и Россолимо, живот немного втягивается на вдохе (парез диафрагмы), чувствительность и координация не нарушены. Диагноз: болезнь мотонейрона, БАС, бульбарный дебют с трипарезом, алиментарная и дыхательная недостаточность, лобно-височные когнитивные экзекуторно-семантические нарушения.
Семейный анамнез. Мать больной также страдала бульбарным дебютом БАС с нарушениями речи, глотания, похуданием, слабостью в конечностях, умерла в возрасте 71 года. Больная имеет двух здоровых детей, сына 44 лет и дочь 39 лет.
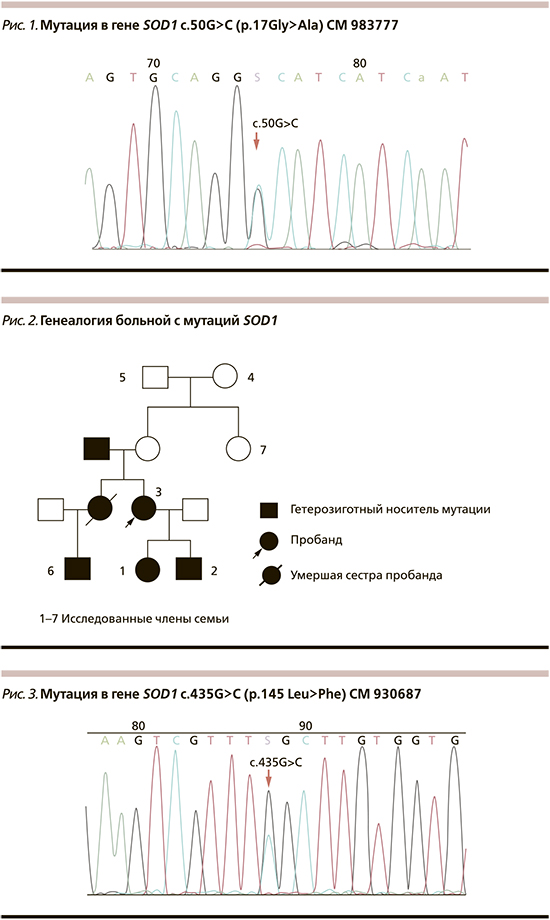
У больной и ее здорового сына выявлена мутация в гене ANG в гетеро-зиготном состоянии c.208 A>G (p.70 Ile>Val) CM 060832 NM_001145.4 (рис. 4). Мутаций в генах SOD1, FUS, VAPB, TARDBP не выявлено. Ген C9orf72 не исследовался. Гены SOD1 и VAPB у пациентов исследовались в Университете Умеа (Швеция), прочие — в России.
Клинический случай 4
Больной Г. 62 лет проживает в Ижевске. В течение полугода отмечает слабость в плечах, ощущение подергиваний. При обследовании на МРТ шейного отдела позвоночника выявлена грыжа С5-6 и С6-7 до 3 мм. В неврологическом статусе когнитивных симптомов нет, черепная иннервация в норме, парез в дельтовидных мышцах и бицепсах до 2—3,5 баллов больше справа, атрофии и фасцикуляции мышц верхнего плечевого пояса, сухожильные рефлексы с рук снижены, с ног — средней живости, патологические кистевые рефлексы, чувствительных, координаторных и тазовых нарушений нет. При игольчатой ЭМГ выявляется спонтанная активность мышечных волокон и двигательных единиц в правой дельтовидной мышце и правой прямой мышце бедра с выраженным повышением длительностей и амплитуд потенциалов двигательных единиц. Диагноз: болезнь мотонейрона, БАС, шейный дебют, синдром Вульпиана-Бернардта, сегментарноядерный вариант, быстрое прогрессирование.
На приеме больной был вместе с братом, который настаивал на сокрытии диагноза.
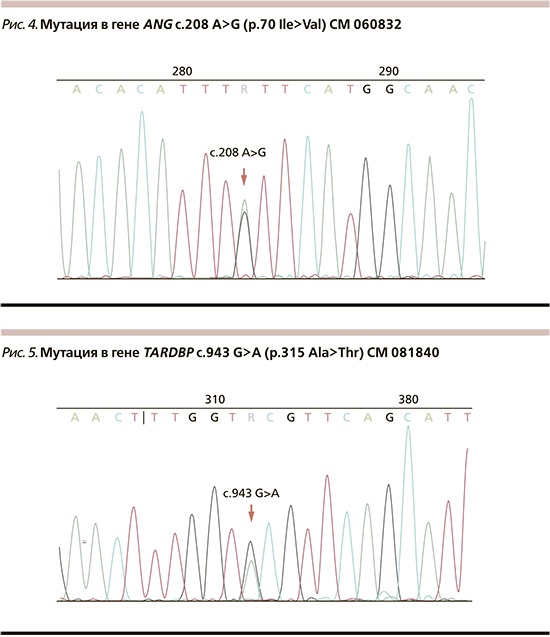
У больного выявлена мутация гена TARDBP с.943 G>A (p.315 Ala>Thr) CM 081840 NM_007375.3 в гетерозиготном состоянии (рис. 5).
Кроме того, были обследованы больная Д. 66 лет с бульбарным дебютом БАС и ее сестра А. 65 лет с поясничным дебютом БАС. У обеих выявлена генетрозиготная мутация в гене TABDRP G 298 S.
Клинический случай 5
Больная О. 64 лет в течение года не предъявляла жалоб, однако ее родные замечали забывчивость, инертность, нарушения самообслуживания, неоднократно терялась на улице, изредка проявляла агрессивность при критике. Также родственники заметили у больной смазанность речи и похудание правой кисти.
Семейный анамнез: ранее случаев подобного заболевания в семье отмечено не было. Родители пациентки были здоровыми и умерли в возрасте 83 и 85 лет. У пациентки две дочери и один сын.
При осмотре в неврологическом статусе: логранк по шкале лобно-височной деменции -0,35 (тяжелые нарушения). Больная дезориентирована во времени, ориентирована на месте, не может нарисовать часы, скопировать кубик, выполнить тест на программирование. Узнавание не нарушено. Слухоречевая память снижена. Нейродинамические нарушения. При тесте на обобщение указывает различия. Оценка по Монреальской шкале 10 баллов (тяжелые нарушения). Глазодвигательных нарушений нет. Жевание не нарушено, мимика не нарушена. Легкая дизартрия и дисфагия. Язык с фасцикуляциями без атрофий. Парезов мышц шеи нет. Парез сгибателей и разгибателей правой кисти до 3 баллов с атрофией первой дорсальной межкостной мышцы. Сухожильные рефлексы низкие, мышечный тонус не изменен. Патологических рефлексов нет. Чувствительность не нарушена. Тесты на координацию, спирографический маневр не выполняет из-за нарушения понимания команд. Диагноз: боковой амиотрофический склероз в сочетании с лобно-височной деменцией.
У пациентки выявлено увеличение числа GGGGCC-повторов в гене C9orf72 (рис. 6). Аналогичная мутация выявлена у одной из дочерей пациентки, у сына не выявлена. Вторая дочь от исследования отказалась.
Обсуждение
Мутация G17A гена SOD1 была впервые описана в португальской, затем в японской семье больных БАС [23]. Клинические детали не сообщаются. Тем не менее следует отметить, что две пациентки с БАС, описанные в настоящей работе, заболели в возрасте около 40 лет, и у них отмечен поясничный дебют БАС с продолжительностью жизни порядка 4—5 лет.
Мутация гена SOD1 L144F описана в первые годы обнаружения мутаций гена SOD1 при БАС. Впоследствии P. Corcia et al. описали семью с тремя пациентами с атипичным респираторным дебютом БАС и этой мутацией гена SOD1 [24]. Интересно отметить, что мы поставили больной по результатам декремент-теста на ЭМГ сочетанную миастению, у больной имела место вероятная тимома переднего средостения. Похожий случай описан в обзоре R. Bedlack по препарату луназин в 2014 г. у некоего мистера Мак Даффа, у которого за несколько лет до развития БАС имела место подтвержденная миастения, а впоследствии он заболел шейным дебютом и сегментарноядерным вариантом БАС, как и наша больная, с улучшением симптоматики после применения препарата луназин [20]. Больная наблюдалась в рамках Благотворительного фонда помощи больным БАС им. Н.И. Левицкой, пользовалась прибором неинвазивной вентиляции легких. От луназина, который пациентка принимала в течение 6 месяцев, улучшений не было. Следует также отметить на указание на БАС в семейном анамнезе больной: мать больной умерла от поперхивания (дебют неясен), бабушка страдала нарушениями ходьбы, движений рук и дыхательными нарушениями, т.е., вероятно, поясничным дебютом БАС. Следовательно, можно сделать вывод о фенотипической гетерогенности БАС в данной семье, однако респираторный дебют у матери больной сомнителен.
Мутации SOD1 выявлены нами в семейных случаях БАС в 3,3%, в НЦ неврологии РАН частота в 24% случаев [14].
Клиническая картина БАС при мутациях в гене ANG весьма разнообразна. На данные мутации приходится до 1% случаев семейного и спорадического БАС [25, 26]. Тем не менее следует отметить, что обе описанные нами больные БАС с мутацией I70V страдали бульбарным дебютом и заболели в возрасте 65-70 лет, продолжительность жизни составила 1,5-2,0 года. У больной имели место лобно-височные когнитивные нарушения. Мутация ANG выявлена нами в 1,5% семейных случаев БАС, в НЦ неврологии РАН - в 1,05% случаев были спорадическими.
Мутации в гене TARDBP или TDP-43, TAR-ДНК-связывающем белке вызывают случаи как семейного, так и спорадического БАС, ассоциированных с наличием TARDBP-позитивных убиквитинированных/тау-негативных включений, могут приводить к развитию случаев БАС с деменцией. Ген локализован на хромосоме 1р36.22, наследование мутаций происходит по АД-типу [27]. Мутации этого гена встречаются у 5-6% больных семейным БАС [2, 28, 29]. Характерны более раннее начало болезни, шейный дебют, медленное прогрессирование [30]. У некоторых больных развивается лобно-височная деменция. Описаны случаи лобно-височной деменции без БАС, ассоциированные с мутациями этого гена [31]. Мутация, описанная нами, была описана ранее P. Corcia et al. в 2012 г.: пациентка заболела шейным дебютом БАС в возрасте 50 лет, впоследствии у нее развилась лобно-височная деменция. У больного, описанного нами, также был шейный дебют БАС, более поздний возраст начала, не было когнитивных нарушений. Дальнейшая судьба данного пациента нам не известна в силу непонимания особенностей заболевания в его случае его родственниками. Мутация этого гена обнаружена в российской популяции впервые, как и мутация G298S в другой семье [14].
Мутация C9orf72 выявлена нами с частотой 1,5% в спорадическом случае, в НЦ неврологии РАН - в 1,8%.
Заключение
Проведенное исследование выявило мутации различных каузативных генов БАС в российской популяции с определенными характерными клиническими особенностями, частота выявленных поражений генов меньше ранее опубликованной для гена SOD1 и совпадает с ранее опубликованными данными для генов ANG и C9orf72. Частота встречаемости мутаций гена C9orf72 значительно ниже таковых в европейских странах. Мутации гена TARDBP выявлена в России впервые. Необходимы дальнейшие исследования по изучению мутаций каузативных генов БАС в России.


