
Введение
В ХХI в. качество оказания медицинской помощи пациентам приобретает новый смысл. В прошлом веке наследственные заболевания в основной своей массе относились к группе неизлечимых болезней. Профилактика таких заболеваний сводилась к расчету риска возможного появления больного ребенка в семье, в 1980-х гг. начала активно развиваться инвазивная пренатальная диагностика, в основном направленная на выявление плодов с хромосомной патологией. С приходом новых лабораторно-технических возможностей одним из важных инструментов в практике врача стали молекулярно-генетическое технологии, позволяющие как найти мутации в отдельном гене, так и выполнить полногеномное секвенирование. Теперь помощь семье стала более адресной, а подтверждающая клинический диагноз диагностика с помощью молекулярно-генетических методов придает уверенность врачу и спокойствие пациенту в правильности диагноза, как следствие – правильным действиям в лечении и профилактике осложнений. Если еще в конце ХХ в. «наследственное заболевание» звучало почти как приговор, то сейчас для многих болезней разработано патогенетическое лечение, а фармацевтические гиганты активно разрабатывают новые препараты для редких (орфанных) болезней. Огромное значение генетическим факторам развития кожных заболеваний придавали такие корифеи дерматологии, как К.Н. Суворова, В.Н. Мордовцев, Г.И. Суколин, и многие другие [1–3]. Наследственные поражения кожи больных описывались как дерматологами, так и врачами других специальностей – офтальмологами, стоматологами, невропатологами, педиатрами, терапевтами и др., поскольку многие наследственные болезни характеризуются сочетанным поражением кожи и других органов и систем [4]. Многие наследственные заболевания с кожными проявлениями традиционно изучаются в равной или даже в большей степени в других разделах медицины.
Термин «дерматогенетика» давно общепринят [1]. Это направление охватывает вопросы изучения наследственных особенностей и аномалий кожи, различных наследственных дерматозов, а также наследственную предрасположенность к приобретенным кожным заболеваниям. В отдельную большую группу заболеваний выделяют факоматозы – генетически, клинически и патогенетически гетерогенную группу, объединенную по принципу локализации патологии в трех системах. Термин «факоматозы» ввел в 1921 г. голландский офтальмолог J. van der Hoeve, описавший опухолевидные образования на сетчатке при туберозном склерозе и предложивший объединить в одну группу врожденные и наследственные заболевания с изменениями кожи в виде гипер- и гипопигментированных пятен, ангиом, нейрофибром, сочетающихся с поражением глаз, нервной системы и нередко – внутренних органов [5]. К факоматозам относят нейрофиброматоз, туберозный склероз, синдром Штурге–Вебера, синдром Гиппеля–Линдау, множественный базоцеллюлярный невус. Другие названия заболеваний этой группы: врожденные нейроэктодермальные дисплазии, гамартобластозы и т.д. Это системные дисплазии с комбинацией опухолевидных пороков развития кожи, глаз, нервной системы. Некоторые авторы считают, что необходимо расширить круг заболеваний, относимых к факоматозам, разделив их на ангиоматозные, эпителиоматозные, эритемокератотические факоматозы и меланофакоматозы.
Одним из самых известных и тяжело текущих факоматозов является нейрофиброматоз I типа (OMIM 162200), синоним: болезнь Реклингхаузена, относящийся к моногенным аутосомно-доминантным заболеваниям, обусловленный мутацией гена NF 1 в 17-й хромосоме. Высокопенетрантное с вариабельной экспрессивностью заболевание. Популяционная частота – 1:3000 новорожденных. Среди генодерматозов доля NF1 составляет около 9% [6]. Необычно высокая частота NF1, по данным Garty et al. (1994), у молодых израильтян – 1,04:1,000 (0,94:1,000 у мужчин и 1,19:1,000 у женщин) [7]. Существует две основные формы: классическая, или периферический нейрофиброматоз, – I тип (85–90%), и билатеральная слуховая, или центральный нейрофиброматоз, – II тип. Хотя нейрофиброматоз I типа был описан еще в 1882 г. немецким врачом Frederich von Recklinghausen, до сих пор у врачей возникают трудности в постановке диагноза, т.к. он очень вариабелен по клиническим проявлениям, даже в рамках одной семьи. Поскольку заболевание может затрагивать все органы системы, на старте клинических проявлений больной может обращаться к разным специалистам. Сложность заключается еще в том, что процесс носит прогрессирующий характер, а усугубить его могут даже обычные внешние факторы, например солнечная инсоляция [8].
Регионы России различаются по этническому и национальному составу, климатогеографическим условиям, солнечной инсоляции. Следовательно, имеются различия в распространенности этой наследственной патологии. В Ростовской области распространенность нейрофиброматоза I типа составляет 1:9212 [9], в Республике Мордовия – 1:10 842 [10], в Башкортастане – 1:21 276 [11].
Цель работы: изучить распространенность заболевания, особенности клинических проявлений нейрофиброматоза I типа в популяции Новосибирской области, включая Новосибирск.
Методы
Ретроспективный анализ карт пробандов клинического отделения медико-генетического отдела (далее МГО) ГБУЗ НСО «ГКБ № 1» за 10 лет (с 2010 по 2020 г.) с установленным клиническим диагнозом «нейрофиброматоз I типа». При его постановке применялись диагностические критерии, рекомендованные Международным комитетом экспертов по нейрофиброматозу: не менее 5 пигментных пятен цвета кофе с молоком диаметром более 5 мм у детей допубертатного возраста и не менее 6 пятен диаметром более 15 мм в постпубертатном возрасте, 2 и более нейрофибромы любого типа или I плексиформная нейрофиброма; веснушчатость в подмышечных и паховых складках, дисплазия крыла клиновидной кости или врожденное истончение кортикального слоя длинных костей с псевдоартрозом или без него; глиома зрительного нерва; два и более узелков Лиша (гамартомы на радужной оболочке) при исследовании с помощью щелевой лампы; наличие родственников первой степени родства с нейрофиброматозом I по тем же критериям. Наличие не менее 2 из этих признаков позволяет ставить диагноз [12].
По данным Росстата, средняя численность населения в Новосибирской области в 2010 г. составляла 2 664 029, в 2020 г. – 2 798 170, прирост населения за 10 лет составил на 134 141.
Результаты
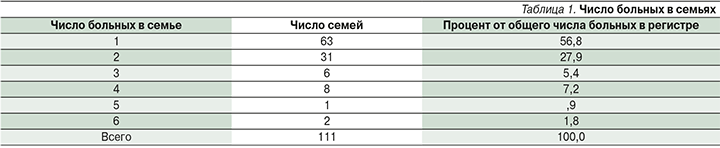
За последние 10 лет в МГО наблюдали 111 пациентов с нейрофиброматозом I типа, с учетом больных родственников (81 человек) всего 192 пациента с нейрофиброматозом I типа. Таким образом, распространенность нейрофиброматоза I типа в Новосибирске и области составила 1:14 500. Спорадических случаев возникновения заболевания было 63, что составило 56,0%, у 48 пробандов имелись родственники 1-й степени родства с установленным диагнозом или он устанавливался впервые на приеме. В большинстве семей помимо пробанда был еще один пациент с NF1 (27,9%; табл. 1). Три больных в семье (включая пробанда) встречались заметно реже. Распределение пробандов по полу оказалось примерно равным – 47% мужчин и 53% женщин.
Обсуждение
Практически не изучена связь влияния средовых факторов как инсоляционная нагрузка с течением степени тяжести клинических проявлений нейрофиброматоза I типа. Сравнительно низкая распространенность заболевания и его медленное прогрессирование могут объясняться особенностями климата: население в осенне-зимне-весеннее время постоянно носит одежду, которая изолирует кожу от солнечных лучей. Это благоприятно для течения нейрофиброматоза. Солнечные лучи при прямом попадании на протяжении длительного времени на кожу больного вызывают резкое ухудшение течения заболевания.
В регистре имеется несколько случаев прогрессирования болезни после длительной инсоляции. Так, у больного С. 27 лет, который обратился за медицинской помощью по поводу аллергической сыпи, при детальном осмотре выявлено пятно цвета кофе с молоком размером 10 на 24 см, расположенное в области 3–5-й реберных дуг справа. Поверхность пятна была покрыта множественными нейрофибромами диаметром 0,1–0,2 см. Данное образование появилось у больного в 5 лет. Дерматолог рекомендовал принимать солнечные ванны для равномерного загара, что больной и выполнял 3–4 летних сезона – до тех пор, пока не обратил внимания на то, что пятно начало увеличиваться в размере, а поверхность его стала неровной. После этого больной к врачам не обращался. При сборе семейного анамнеза оказалось, что у его деда по материнской линии были похожие пятна, но меньшего размера, а умер он после удаления меланоцитарного невуса в области лба (из справки о смерти больного). У матери пробанда при осмотре множественные пятна типа веснушек. С учетом клинических проявлений и родословной больного был поставлен диагноз «нейрофиброматоз I типа».
У другой больной, Г. 42 лет, в возрасте 7–8 лет появились пятна кофейного цвета на груди и спине. В течение последующих 6 лет девочка проводила летние каникулы на Черноморском побережье. После второго сезона на груди появились опухолевидные образования, с которыми она к врачу не обращалась. Девочка продолжала отдыхать под южным солнцем. Число нейрофибром нарастало. После обращения к генетику ей был выставлен диагноз «нейрофиброматоз I типа». Процесс неуклонно прогрессировал. Число нейрофибром увеличивалось на несколько десятков ежегодно. К 42 годам на коже больной насчитывалось более 400 нейрофибром (диаметром от 0,5 до 2,0 см), множественные лентиго, появились признаки поражения внутренних органов, изменения психики.
В пользу климатического влияния на экспрессивность нейрофиброматоза – преобладание слабой и средней степеней тяжести его клинических проявлений в нашем регионе (табл. 2). При нейрофиброматозе I типа степень выраженности клинических проявлений оценивалась по разработанным для данного исследования критериям следующим образом: слабая – при наличии у больного не менее двух из перечисленных признаков (не менее пяти пигментных пятен цвета кофе с молоком диаметром более 5 мм у детей до пубертатного возраста, не менее 6 пятен диаметром более 15 мм в постпубертатном возрасте, от двух до десяти нейрофибром любого типа, веснушчатость в подмышечных или паховых складках, наличие у родственников первой степени родства нейрофиброматоза I по тем же критериям); средняя – пятна диаметром более 15 мм и нейрофибромы любого типа диаметром до 15 мм не более 100 штук; выраженная – любые кожные проявления нейрофиброматоза с поражением внутренних органов или при количестве нейрофибром, превышающем 100 штук.

Слабая степень выраженности клинических проявлений встречалась у 38,7% пациентов. Поскольку проявления при данной степени мало выражены, эти случаи могут вызывать трудности со стороны узких специалистов при постановке диагноза. При анализе диагноза направления таких пациентов на консультацию в МГО только у 8 из 43 пациентов был зафиксирован диагноз «нейрофиброматоз I типа», остальные пациенты были направлены для исключения наследственной патологии. Средняя степень выраженности преобладала и встречалась у 46,8% пациентов. Диагноз «нейрофиброматоз I типа» в направлении прослеживался у 29 пациентов из 52, у 13 было зафиксировано: исключить «факоматоз», у 10 – исключить наследственную патологию. Выраженная степень была у 16 пациентов, что составило 14,4%, у всех пациентов диагноз направления «нейрофиброматоз I типа». Результаты этого анализа свидетельствуют о малой настороженности врачей в отношении данного диагноза при слабой и средней степеней выраженности нейрофиброматоза I типа, что может стать причиной неконтролируемого прогрессирования процесса.
Наиболее ранний признак во всех случаях – полиморфные по размерам пятна цвета кофе с молоком с первичной локализацией в подмышечных впадинах, реже в паховых складках на шее, под молочными железами.
В целом они выявлялись почти в 100% случаев. У большинства они были с рождения или появлялись в течение 1-го года жизни, у 12% больных – до 10 и только у 2% – после 10 лет, что совпадает с данными В.В. Мордовцева [2].
У нескольких больных пятна появлялись в местах расчесов и трения одежды. Только в одном случае на пигментном пятне имелись мелкие нейрофибромы. Изменения кожи имели внутри- и межсемейные различия по времени появления клинических симптомов и их вариабельности. Помимо перечисленных симптомов, у всех пациентов при прикосновении к коже ее можно было охарактеризовать как «бархатистую».
Второй по частоте встречаемости признак заболевания – нейрофибромы, разные по размерам (от 2–3 мм до нескольких сантиметров), распространяющиеся по ходу периферических нервов преимущественно на туловище, руках, лице, у 13,5% больных единичные, у 20,7% – множественные. Процент больных нейрофибромами зависит от возраста.
Нейрофибромы появляются значительно позже пятен, обычно в 12–17 лет. К 20 годам 50% больных имеют нейрофибромы. Однако плексиформные нейрофибромы возникали в раннем детстве, с течением времени их число также часто возрастало. Но даже при тяжелой форме нейрофиброматоза I типа не наблюдалось гигантских нейрофибром, описанных в литературе. У 28% больных имели место изменения со стороны опорно-двигательного аппарата (сколиоз, кифоз, плоскостопие). У 6 пациентов обнаружена глиома зрительного нерва, узелки Лиша – у 2.
Обобщая вышесказанное, можно предположить, что наш регион благоприятно влияет на течение нейрофиброматоза. Возможно, больные с малыми признаками, такими как лентиго в подмышечных впадинах, даже не знают о своем заболевании. Вполне вероятно, что это и есть одна из главных причин более низкой выявляемости нейрофиброматоза I типа в нашей популяции.
Хочется подчеркнуть, что речь идет не о частоте встречаемости патологического гена в нашей популяции, а именно о частоте заболевания. Частота мутаций в гене может не отличаться от таковой в других регионах, но вот процент экспрессивности благодаря благоприятным климатогеографическим особенностям региона может быть намного меньше, чем в районах с интенсивной инсоляцией (Израиль – 1:1000, Италия – 1:3500). Хотя, безусловно, некоторая часть различий может объясняться этническими особенностями.
Заключение
Распространенность нейрофиброматоза I типа в нашей популяции составила 1:14 500, что значительно ниже, чем в других популяциях. Особенность клинических проявлений заключается в том, что на территории НСО слабая степень выраженности клинических проявлений отмечена у 38,7%, а выраженная только у 14,4%, что затрудняет раннюю диагностику заболевания. Нейрофиброматоз – это полисистемное заболевание, требующее системного подхода к диагностике и коррекции.


