Введение
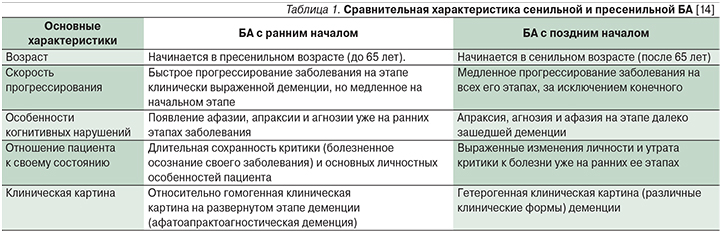
Болезнь Альцгеймера (БА) – нейродегенеративное заболевание, проявляющееся прогрессивным снижением памяти и других когнитивных функций вследствие гибели нейронов коры больших полушарий [1]. БА с ранним началом (до 65 лет) составляет 1–5% от всех случаев БА [2]. В США насчитывают примерно 200 тыс. пациентов с ранней формой БА, что как раз составляет около 4% от 5,7 млн больных БА [3]. В российских исследованиях данные о заболеваемости пациентов с БА с ранним началом отсутствуют. По данным Росстата, смертность от БА с ранним началом за 2012 г. у пациентов в возрасте 35–44, 45–54 и 55–64 года составила 0,005, 0,038 и 0,330 на 100 тыс. населения соответственно [4]. Актуальность проблемы заключается не только в том, что ранняя форма хуже диагностируется в связи с редкой встречаемостью, протекает более быстро и агрессивно, а также длительно осознается пациентом по сравнению с БА с поздним началом (табл. 1), но и в том, что пациентами становятся лица трудоспособного возраста, что способствует формированию разнообразных психологических, социальных и экономических проблем [5].
В настоящее время понятие БА с ранним началом расширено и дополнено. БА с ранним началом может иметь как семейную, так и спорадическую формы [6]. Причем около половины случаев семейной формы БА с ранним началом приходится на аутосомно-доминантную форму [7]. Семейная форма БА может относиться к заболеванию не только с ранним, но и с поздним началом (после 65 лет). Причем часть случаев семейной формы БА с поздним началом приходится также на аутосомно-доминантную форму [8]. Таким образом, существует несколько сходных, в определенной степени перекрывающихся состояний: семейная форма БА (FAD – Familial Alzheimer’s Disease) [9], БА с ранним началом (EOAD – Early-Onset Alzheimer’s Disease) [10], семейная форма БА с ранним началом (EOFAD – Early-onset familial Alzheimer’s disease) [11], аутосомно-доминантная БА (ADAD – autosomal dominant Alzheimer’s disease) [12] и аутосомно-доминантная форма БА с ранним началом (ADEOAD – early-onset autosomal dominant Alzheimer’s disease) [13].
В настоящей статье речь пойдет о семейной форме БА с ранним началом, включая аутосомно-доминантную форму.
БА с ранним началом обычно начинается в возрасте от 35 до 65 лет. Средний возраст больных составляет 54–56 лет, средняя продолжительность жизни после установления диагноза – 8–10 [15]. Заболевание сопровождается прогрессирующим нарушением памяти, интеллектуальной деятельности и высших корковых функций, приводит к развитию тотальной деменции с синдромами афазии, апраксии и агнозии [16]. Возможно проявление зрительной дисфункции и дискалькулии [17].
Также, по данным И.В. Колыхалова (2016), у 60% пациентов на том или ином этапе развития заболевания имеют место поведенческие и психопатологические расстройства следующих групп:
- Психотические расстройства (бредовые – 31%, зрительные и слуховые галлюцинации – 13 и 10% соответственно);
- Аффективные расстройства (депрессия – более 50%, тревога – от 24 до 65%, апатия – от 19 до 76%);
- Поведенческие нарушения (агрессия – 6%, двигательное беспокойство [аберрантное моторное поведение] – 12–84%) [18].
Патоморфологическая картина характеризуется церебральной атрофией с уменьшением объема и массы мозга, атрофией извилин коры, c расширением корковых борозд и желудочковой системы. При микроскопическом исследовании отмечается массивная утрата нейронов коры мозга, гиппокампа, а также базального ядра Мейнерта и голубого пятна ствола мозга. У сохранившихся нейронов выявляется дегенерация дендритов [19].
В связи с тем что семейная форма БА может дебютировать в разное время (быть БА с ранним или поздним началом), исследователи уделяют большое внимание этиологическим факторам развития заболевания. Считается, что чем позже наступает БА, тем больше доминируют факторы старения и окружающей среды (лентивирусы, дефекты энергетического метаболизма, недостаточность нейротрофических факторов, эксайтотоксичность, митохондриальные дефекты, нейротоксичность микроэлементов и окислительный стресс [20]) и тем меньше влияет генетическая предрасположенность. А чем раньше возраст начала, тем БА будет больше определяться детерминированной мутацией в одном гене [21].
Генетические аспекты БА с ранним началом
БА с ранним началом – заболевание практически с полной генетической детерминированностью, наследуемость составляет от 92 до 100% [22].
У 35–60% пациентов с БА с ранним началом обнаруживается не менее одного родственника первой степени родства с данным заболеванием (т.е. семейная форма БА с ранним началом), причем у 10–15% пациентов с семейной БА с ранним началом отмечается аутосомно-доминантный тип наследования [23]. Аутосомно-доминантная форма БА с ранним началом составляет 0,5% от всех случаев БА [9].
Почти у 50% родственников пациентов с симптомами аутосомно-доминантной формы БА с ранним началом выявляются мутации в следующих генах [24] (табл. 2):
- ген белка – предшественника амилоида (APP – amyloid precursor protein) локализован на 21-й хромосоме; тип наследования аутосомно-доминантный с полной пенетрантностью;
- ген белка пресенилина 1 (PSEN1 – presenilin protein 1) локализован на 14-й хромосоме; тип наследования аутосомно-доминантный с неполной пенетрантностью;
- ген белка пресенилина 2 (PSEN2 – presenilin protein 2), локализующегося на 1-й хромосоме; тип наследования аутосомно-доминантный с неполной пенетрантностью.
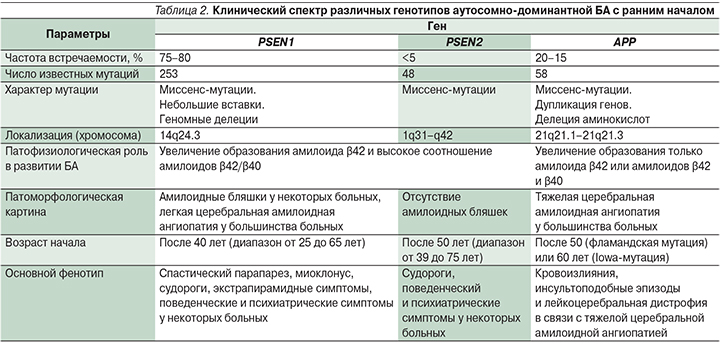
Мутации гена PSEN1 ответственны за 75–80% случаев развития БА с ранним началом, мутации генов APP и PSEN2 – за 20–15% и <5% случаев соответственно [24].
Большинство мутаций пресенелина – однонуклеотидные замены, однако также описаны мелкие делеции и инсерции. Мутации PSEN1 и PSEN2 вызывают изменение аминокислотной последовательности белка с кластеризацией трансмембранных доменов и гидрофильных петель; 75% мутаций гена APP – это миссенс-мутации, расположенные в пределах 16-го и 17-го экзонов в сайте расщепления β- и γ-секретаз. Оставшиеся 25% мутаций гена APP представлены дупликациями. Мутации одного из этих трех генов в настоящее время служат единственными факторами, позволяющими достоверно прогнозировать развитие БА.
К настоящему времени выявлено около 253 различных патогенных мутаций гена PSEN1, 48 мутаций гена PSEN2 и 58 – (или удвоение) гена APP при аутосомно-доминантной форме БА с ранним началом [25]. Почти половина семей, удовлетворяющих критериям аутосомно-доминантной формы БА с ранним началом, служат носителем одной из этих мутаций. Генетический дефект, лежащий в основе возникновения остальных 50% случаев аутосомно-доминантной БА с ранним началом, остается неизвестным, что указывает на наличие других генетических и/или этиологических факторов [26].
Если говорить о спорадической, а не семейной (аутосомно-доминантной) форме БА с ранним началом, то она составляет 50% от всех случаев БА с ранним началом [27]. Такие пациенты не имеют семейного анамнеза, но после генетического тестирования у них могут быть выявлены известные мутации в одном из трех доминирующих (первичных) генов – PSEN1, PSEN2 или APP. Также может быть обнаружена неизвестная мутация в одном из этих трех генов. Однако не всегда возможно определить причину, т.к. мутация может произойти в гене, роль которого в возникновении БА еще не доказана, кроме того, причина может быть связана с эпигенетическими нарушениями [28].
Помимо трех рассмотренных выше первичных генов существуют и вторичные. Наиболее изученный – ген ApoE (apolipoprotein E), который играет жизненно важную роль в транспортировке липидов и холестерина, опосредует синаптическую пластичность, синаптогенез и воспаление [29]. Ген ApoE расположен в хромосоме 19 и состоит из 3 основных изоформ: e2, e3 и e4. Изоформа e4 гена ApoE служит фактором риска БА [30]. Выявление двух аллелей ApoE4 (гомозиготный носитель) приводит к увеличению вероятности развития БА в 10–12, а одной аллели (гетерозиготный носитель) – в 3 раза, но на основе APP или PSEN2 (но не PSEN1) совместно с носительством гена ApoE4 заболевание наступает в более молодом возрасте [32] по сравнению с пациентами с более нейтральным ApoE3 или защитным ApoE2 (снижает риск развития БА [33]).
Механизм, в результате которого ApoE4 увеличивает риск БА, до конца не изучен. Исследования на животных и человеке демонстрируют, что ApoE может влиять на синтез, разрушение и агрегацию β-амилоида. ApoE4 показывает меньшую эффективность в удалении амилоида β42, в результате чего он избыточно осаждается в нейронах головного мозга [34].
Таким образом, исследование пациентов с аутосомно-доминантной формой БА с ранним началом имеет огромное значение для понимания механизмов прогрессирования заболевания от состояния «отсутствия когнитивных нарушений» до «деменции» [35]. Кроме того, в связи почти со 100%-ной пенетрантностью лонгитудинальное наблюдательное исследование пациентов с мутациями, приводящими к аутосомно-доминантной БА с ранним началом, могут позволить проследить клиническую динамику БА от доклинической стадии до фазы умеренного когнитивного снижения (MCI – mild cognitive impairment), а затем деменции [36–38].
Клиническая картина семейной формы БА с ранним началом
Среди типичных клинических симптомов ранней аутосомно-доминантной БА наиболее распространена амнезия. Это характерно для пациентов с мутациями во всех трех генах и дупликацией гена APP. Что касается отдельных мутаций, то среди типичных симптомов при мутации в гене PSEN1 наиболее распространена афазия, среди мутаций в гене PSEN2 и APP – дезориентация, среди дупликаций в гене APP – апраксия. В случае нетипичных клинических проявлений при мутации в генах PSEN1 и APP преобладает миоклонус, в гене PSEN2 и при дупликации гена APP – эпилептические приступы.
Так, в исследовании Y.F. Shea et al. (2016) было выявлено, что амнезия наблюдалась у 84,2% пациентов с мутациями в гене PSEN1, 87,9% – в гене PSEN2, 81,7% – в гене APP и 85,7% случаев при дупликации гена APP. Апраксия наблюдалась у 14,6%, 24,2; 18,3 и 42,8% пациентов при мутации в генах PSEN1, PSEN2, APP и дупликации APP соответственно; афазия – у 30,6%, 21,2; 20,7 и 28,6% соответственно; нарушение ориентации в пространстве – у 19,0%, 36,3; 29,3 и 35,7% соответственно; эпилептические приступы – у 29,4%, 15,2; 15,9 и 66,7% пациентов соответственно [9].
Патофизиология БА с ранним началом
Хотя теория амилоидного каскада – всего лишь один из нескольких возможных механизмов, объясняющих патогенез БА (например, тау-белок, гипотеза оксидантного стресса, воспалительная теория), только отложением амилоида нельзя объяснить возникновение БА. Взаимосвязь генов PSEN1/PSEN2/APP с синтезом и метаболизмом амилоида свидетельствует о том, что теория амилоидного каскада играет более важную роль в патогенезе аутосомно-доминантной БА с ранним началом по сравнению со спорадическими случаями БА [39, 40]. В соответствии с теорией амилоидного каскада БА cлужит следствием дисбаланса между образованием и клиренсом амилоида β [41]. При аутосомно-доминантной БА с ранним началом мутации PSEN1, PSEN2 или APP объединены общим патогенетическим механизмом, приводящим к увеличению образования амилоида β42 [42]. В норме белок APP расщепляется ферментом α-секретазой на одинаковые по величине полипептиды, которые не являются патогенными. При миссенс-мутациях или удвоениях гена APP соответствующий белок расщепляется β- и γ-секретазами на различные по длине фрагменты. При этом длинные фрагменты (β42) нера-створимы и поэтому откладываются в паренхиме головного мозга и стенках церебральных сосудов [43]. Мутации гена APP, расположенные рядом с сайтом расщепления β-секретаз, приводят к повышению протеолиза β-сайта и повышению уровня как амилоида β40 (короткой цепи), так и амилоида β42 [44]. Напротив, мутации, расположенные в области сайта расщепления γ-секретазы, повышают образование амилоида β42 [45].
Пресенилины являются частью γ-секретазного комплекса и функционально вовлечены в протеолитическое расщепление белка – предшественника амилоида [43, 46]. Мутации генов PSEN1 или PSEN2 изменяют скорость расщепления белка – предшественника амилоида, что приводит к увеличению скорости образования амилоида β42 с высоким β42/β40 (длинная/короткая цепь) соотношением [43, 46]. Предполагается, что усиление токсического действия амилоида β42 запускает каскад патологических процессов, включая тау-гиперфосфорилирование, образование нейрофибриллярных клубочков, воспаление нервной ткани, потерю синаптических соединений и гибель нейронов, с возникающей в результате клинической картины деменции как проявление нейродегенеративных процессов [41].
Диагностика семейной формы БА с ранним началом
Для идентификации новых генов, связанных с семейной формой БА с ранним началом, нужны полные родословные семей пациентов, что не всегда возможно, т.к. пациент может не знать о наличии болезни у родственников первой степени. Ведь смерть человека от других причин может наступить раньше, чем клиническое проявление болезни, как правило, запаздывающее на 10–15 лет [47]. Постановка правильного диагноза в случае семейной формы заболевания крайне важна, т.к. далее необходимо предложить родственникам пациента пройти генетическое тестирование и при положительном результате начать раннюю профилактику, а затем своевременное лечение.
Сложность диагностики семейной БА с ранним началом заключается в ее вариабельности (см. рисунок), т.к., например, у одного члена семьи она может проявиться в возрасте 30 лет, у другого – 60. Кроме того, у разных членов одной семьи заболевание может проявляться разными клиническими симптомами: у одного пациента – парез и постепенный паралич нижних конечностей и лишь незначительные когнитивные нарушения, у другого – классическая симптоматика БА [48]. Такие различия служат результатом мутации во вторичных генах, которые предположительно изменяют эффект мутации в первичных генах БА, уменьшая или увеличивая время начала заболевания или создавая другую комбинацию симптомов [49].
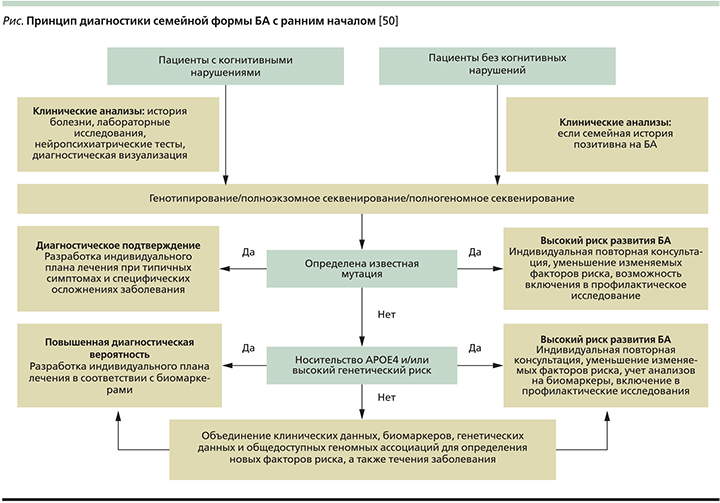
Основные аспекты диагностики семейной формы БА с ранним началом:
- генетическое тестирование, в т.ч. полногеномный поиск ассоциаций [51];
- магнитно-резонансная томография головного мозга (атрофия медиальных отделов височной доли, миндалины, гиппокампа, задней части поясной извилины) [1];
- позитронно-эмиссионная томография – ПЭТ (снижение метаболизма глюкозы в вышеуказанных отделах) [1];
- новый метод прижизненной визуализации β-амилоида в головном мозге с помощью ПЭТ с применением радиоактивно-меченого аналога флуоресцентной краски тиофлавина T (PIB – Pittsburgh compound B) [52] (накопление амилоидного белка в церебральных структурах до развития симптомов нарушения памяти);
- исследование цереброспинальной жидкости – ЦСЖ (увеличение концентрации тау-протеина и уменьшение концентрации β-амилоида) [53]. Концентрация β-амилоида в ЦСЖ у здоровых людей выше, чем у пациентов с БА, т.к. при развитии БА происходит накопление амилоидного белка в тканях головного мозга, а его концентрация в ЦСЖ снижается. Концентрация тау-протеина в ЦСЖ повышается вследствие гибели нейронов церебральных структур (то же можно видеть при инсульте, энцефалите, генерализованном эпилептическом припадке и т.д.) [1].
Медикаментозные методы лечения семейной формы БА с ранним началом
На сегодняшний день из-за низкого процента пациентов с семейной формой БА с ранним началом нет широкомасштабных клинических испытаний препаратов для данной категории больных. Поскольку патогенез и клинические признаки у пациентов с семейной формой БА с ранним началом схожи с таковыми при спорадической форме, общепринятой практикой является использование для лечения семейной формы симптоматических препаратов, эффективность которых доказана для спорадической формы БА с ранним началом.
Базисная терапия. К препаратам базисной симптоматической терапии при БА относят два класса лекарственных препаратов: ингибиторы центральной ацетилхолинэстеразы (ривастигмин, донепезил, галантамин и т.д.) и антагонисты N-метил D-аспартат (NMDA)-рецепторов (мематина гидрохлорид) [1].
Ингибиторы ацетилхолинэстеразы необходимы из-за имеющегося дефицита ацетилхолина, т.к. ферменты, участвующие в его метаболизме, вовлекаются в амилоидогенез [47]. Лечение препаратами данной группы должно быть длительным и начинаться с высоких доз. Критерием эффективности терапии считается клинически доказанное улучшение когнитивных функций или отсутствие ухудшения в течение по крайней мере трех месяцев [54].
К дополнительным критериям эффективности относятся улучшение адаптации пациента в повседневной жизни, уменьшение выраженности эмоциональных и поведенческих расстройств, снижение дозировки или отказ от сопутствующих психотропных препаратов. В отсутствие терапевтического эффекта одного препарата его можно заменить на другой той же группы [55]. Распространенными побочными эффектами являются желудочно-кишечные симптомы (тошнота, рвота, ощущение вздутия живота, снижение аппетита, потеря массы тела). С целью их предотвращения рекомендуют прием пролонгированных пероральных или трансдермальных форм препаратов [56].
Мемантина гидрохлорид оказывает влияние на NMDA-рецепторы, стабилизируя мембрану нейрона и уменьшая эксайтотоксичность [57]. Также данный препарат опосредованно влияет на состояние дофаминергических и ацетилхолинергических церебральных структур, повышая содержание ацетилхолина и дофамина, соответственно, уменьшая выраженность как преимущественно дофаминергических (скорость психических процессов), так и ацетилхолинергических (уровень внимания, способность к длительному удержанию внимания, память) когнитивных симптомов [58]. Побочные эффекты при приеме препарата, как правило, минимальны [59].
При наличии психических и поведенческих расстройств используют психотропные препараты: нейролептики, антидепрессанты, бензодиазепины, противосудорожные средства [60]. Однако серьезным недостатком традиционных (типичных) нейролептиков при лечении пациентов с БА считается высокий риск развития необратимых или малообратимых экстрапирамидных симптомов, а также периферических и центральных антихолинергических побочных эффектов. По этой причине нейролептики следует использовать только для лечения тяжелых расстройств. Также рекомендуется назначение препаратов, не обладающих (или обладающих минимальными) антихолинергическими свойствами [15].
Специфические методы лечения (иммунотерапия). В настоящее время созданы препараты, основанные на моно- и поликлональных антителах, аффинных к амилоиду β42. При использовании моноклональных антител амилоид β42 связывается в единый с ним комплекс и затем выводится из вещества головного мозга. Исследования последних лет позволили создать препараты, обладающие достаточной эффективностью, высокой аффинностью к β-амилоиду и низкой токсичностью [61]. В настоящее время наиболее известны три препарата: бапинейзумаб, соланезумаб и гантенерумаб [62]. Но достоверным клиническим эффектом обладает только гантенерумаб – первый препарат, полностью созданный на основе человеческих анти-Аβ-моноклональных антител [63]. Его действие основано на проникновении через гематоэнцефалический барьер с последующей активацией микроглии, запуском микроглиально-опосредованного фагоцитоза и как следствие – связыванием и удалением β-амилоида из вещества головного мозга. Согласно результатам двойного слепого рандомизированного плацебо-контролируемого исследования, препарат эффективен для пациентов с легкой и умеренной деменцией при БА за счет достоверного уменьшения плотности церебральных амилоидных отложений в основной группе [64].
Заключение
БА – одна из наиболее распространенных форм деменции, затрагивающей более 26 млн человек во всем мире. По сравнению со спорадической БА семейная форма БА с ранним началом имеет некоторые отличительные особенности: ранний возраст начала, положительная семейная история, множество некогнитивных неврологических симптомов и более агрессивное течение заболевания. Существует выраженная фенотипическая гетерогенность среди различных мутаций семейной формы БА с ранним началом. Диагностика данной формы БА должна основываться на клиническом и семейном анамнезе, неврологических симптомах и обследованиях, особенностях биомаркеров, а также генотипировании. Новые терапевтические агенты, нацеленные на выведение β-амилоида, могут помочь в лечении данной категории пациентов.
