
Множественная миелома: общие положения
Несмотря на то что множественная миелома (ММ) относится к наиболее изученным гемобластозам, этиология и патогенез данного заболевания до настоящего времени вызывают споры среди исследователей [1]. ММ составляет 1% всех видов злокачественных опухолей и около 10% всех гематологических злокачественных новообразований. Частота заболеваемости в Европе составляет 4,5–6,0 случаев на 100 тыс. населения в год, средний возраст при постановке диагноза – 72 года; смертность составляет 4,1 случая на 100 тыс. населения в год [2, 3]. Социальная значимость ММ обусловлена неуклонным прогрессированием заболевания, значительной вариабельностью клинических проявлений, наличием большого числа осложнений, а также неудовлетворительным качеством жизни при выживаемости пациентов не более 3–5 лет [1].
Точная этиология ММ еще не установлена. Предположительно это генетические факторы, моноклональная гаммапатия неопределенного значения (МГНЗ или MGUS), ионизирующее излучение, хроническое воспаление и инфекция, экологические или профессиональные причины [4]. Почти у всех пациентов ММ начинает развиваться от бессимптомной предзлокачественной стадии – MGUS, которая прогрессирует до MM со скоростью 1% в год. У некоторых пациентов выявляется т.н. тлеющая MM (SMM – smouldering multiple myeloma), которая приводит к ММ в первые годы после постановки диагноза [2, 5].
Клиническая картина ММ может варьироваться от бессимптомной до тяжелой формы с осложнениями, требующими экстренного лечения. К признакам ММ относятся боли в костях, патологические переломы, компрессия спинного мозга вследствие патологических переломов, слабость, недомогание, кровотечения, анемия, инфекция (часто пневмококковая), гиперкальциемия, почечная недостаточность, невропатии.
Остеолизис и остеопороз, патологические переломы служат проявлением костных изменений при ММ. Патогенез обусловлен секрецией миеломными клетками остеокластактивирующих факторов. Возросшее число активных остеокластов приводит к полному разрушению костной матрицы. По причине дисбаланса костного ремоделирования нарушается резорбция и формирование костной ткани. Некоторые исследователи утверждают, что на сегодняшний день нет эффективных механизмов не только лечения ММ, но и превенции костных осложнений данного заболевания, что также обусловливает актуальность данной проблемы [1].
Диагностические подходы
Диагностика ММ, согласно рекомендациям ESMO-2017, основана на следующих тестах [2, 6, 7]:
- обнаружение и оценка парапротеинов или моноклонального компонента (МС) с помощью электрофореза в сыворотке и/или моче (концентрат 24-часового сбора мочи); количественная оценка иммуноглобулина G (IgG), IgA и IgM; исследование тяжелых и легких цепей с помощью иммунофиксации и измерение легких цепей в сыворотке (FLC – Freelite test);
- оценка инфильтрации плазматическими клетками костного мозга: аспирация и/или биопсия являются стандартными для оценки числа и характеристики плазматических клеток в костном мозге. Кроме того, биоптат костного мозга возможно использовать для исследований цитогенетической/флуоресцентной in situ гибридизации (FISH – fluorescence in situ hybridization) на иммунологически распознанных или отсортированных клетках плазмы, также возможно проведение иммунофенотипических и молекулярных исследований;
- оценка поражения костей: низкодозовая компьютерная томография всех костей (WBLD-CT – whole-body low-dose computed tomography) является новым стандартом для диагностики остеолизиса. Обычная рентгенография используется, если WBLD-CT недоступна. Магнитно-резонансная (МРТ) визуализация дает точную информацию и рекомендуется в тех случаях, когда предполагается компрессия спинного мозга. МРТ всего тела или МРТ позвоночника и таза может быть использована в соответствии с ее доступностью для оценки остеодеструкции, клеточной инфильтрации, наличия фокальных костных поражений. Позитронно-эмиссионная томография с компьютерной томографией (ПЭТ-КТ) с контрастированием 18F-фтордезоксиглюкозой также может быть использована для оценки костных повреждений;
- полное число клеток крови в костном мозге, сывороточный креатинин, клиренс креатинина и уровень кальция. Применение этих тестов позволяет проводить дифференциальную диагностику между MM, SMM и MGUS [2].
Критерии для постановки диагноза ММ [2, 8]:
1) наличие 10% плазматических клеток в костном мозге и/или наличие плазмоцитомы внекостномозговой локализации;
2) выявление в сыворотке крови моноклонального белка и/или выявление его в моче (за исключением пациентов с несекретирующей [без продукции моноклонального белка] ММ);
3) обнаружение любого из критериев, определяющих миелому:
а) повреждение органов-мишеней (т.н. критерии CRAB: гиперкальциемия более 11,5 мг/дл (>2,65 ммоль/л), почечная недостаточность (креатинин >2 мг/дл или >177мкмоль/л) или клиренс креатинина <40 мл/мин: рассчитывается с помощью формул MDRD или CKD-EPI [5], анемия (гемоглобин <100 г/л или на 20 г/л ниже нормального уровня) или поражения костей (остеолитические поражения, остеопороз или патологические переломы);
б) остеолизис, определяемый по данным КТ.
4) Любые биомаркеры злокачественности:
а) 60% клональных плазменных клеток костного мозга (КМ);
б) соотношение вовлеченных и невовлеченных (связанных и несвязанных) легких цепей в сыворотке выше или равное 100;
в) более одного очагового поражения костей на исследованиях МРТ (каждое очаговое поражение должно быть не менее 5 мм).
Наиболее часто сопровождают ММ такие симптомокомплексы, как амилоидоз, рецидивирующие инфекции чаще двух раз за 1 год, синдром гипервязкости крови.
Одним из наиболее распространенных осложнений ММ является амилоидоз. Частота возникновения амилоидоза при ММ составляет 20% среди всех осложнений. Амилоидоз – группа заболеваний, отличительным признаком которых является накопление в тканях и органах фибриллярного гликопротеида амилоида [9]. В зависимости от локализации процесса разделяют локальные и системные (генерализованные) формы амилоидоза. Диагностика амилоидоза основывается на результатах морфологического исследования тканей пораженного органа (прямая кишка, почки, печень). С целью выявления амилоида обычно применяют окрашивание препаратов ткани красителем конго-красный с последующей микроскопией в поляризованном свете, для дифференциальной диагностики амилоидоза применяют окраску с тиофлавином Т.
Синтез большого количества нестабильных белков-предшественников (специфичны для каждого типа амилоидоза), которые образуют агрегаты с последующим формированием амилоидной фибриллы, является ведущим фактором в патогенезе амилоида. Изменения в первичной структуре белков-предшественников (первичные и вторичные) определяют их амилоидогенность. Воспалительные процессы и опухоли могут служить причиной повышения количества сывороточного амилоида А (SAA) в несколько раз при воспалительных процессах и опухолях [10].
В основу современной классификации амилоидоза положен принцип специфичности основного фибриллярного белка амилоида (см. таблицу). Так как тип амилоида определяет тактику ведения пациента, современная патоморфологическая диагностика амилоидоза включает обнаружение и обязательное типирование амилоида. Наиболее значимым методом типирования амилоида является иммуногистохимическое исследование с определением специфического антитела против АА белка, легких цепей иммуноглобулинов, транстиретина и β2-микроглобулина. Иногда целесообразно применение методов протеомного анализа амилоида.
ММ, как правило, осложняется AL-амилоидозом. В-лимфоцитарная дискразия (формирование аномального клона плазматических или В-клеток в костном мозге, продуцирующего амилоидогенные иммуноглобулины) служит единым этиологическим фактором всех форм AL-амилоидоза. АL-амилоидоз является наиболее распространенным типом системных амилоидов в развитых странах с уровнем заболеваемости 9 случаев на 1 млн человек в год. Средний возраст пациентов с диагнозом составляет 65 лет, а менее 10% пациентов моложе 50 лет [12]. AL-амилоидоз характеризуется системным поражением внутренних органов: сердца, почек, поражением дыхательной, нервной систем, кожи. Печень при данном виде амилоидоза, по данным авторов российских клинических рекомендаций, поражается в 100% случаев. К признакам поражения печени относятся гепатомегалия и холестаз. Характерно повышение активности γ-глутамилтранспептидазы (ГГТП) и щелочной фосфатазы (ЩФ) до 3–4 норм. Данные изменения происходят на фоне сохранной функции печени. Для длительного течения заболевания характерна портальная гипертензия, сочетающаяся с выраженной желтухой. При таком варианте течения возможны следующие осложнения: кровотечение из основных портосистемных шунтов (вены пищевода, геморроидальные вены), снижение функциональной активности органа (из-за накопления амилоида), также развитие печеночной комы [13]. Одним из методов обследования, направленным на оценку функционального резерва печени, является 13С-метацетиновый дыхательный тест, с помощью которого возможно определить минимальные отклонения в активности системы CYP450 печени [14].
Принципы терапии
Основной задачей лечения амилоидоза является терапия заболевания, следствием которого он является, а именно уменьшение количества или элиминация белков-предшественников, чтобы замедлить или на время остановить эволюцию заболевания. При ММ целью лечения является подавление пролиферации клона плазматических клеток, уменьшение продукции легких цепей иммуноглобулинов [15]. Срок лечения составляет не менее года, проведение его затрудняется тем, что достижение положительного эффекта препаратов происходит медленнее, чем прогрессирование болезни. Патогенетическая терапия ММ позволяет уменьшать накопление и отложение в тканях амилоида, улучшая таким образом прогноз пациентов.
В настоящее время в клинической практике применяются противоопухолевые препараты (бортезомиб, леналидомиб, иксазомиб, талидомид, карфилзомиб, элотозумаб, даратомумаб), системные глюкокортикостероиды (преднизолон, дексаметазон), цитостатики (мелфалан, циклофосфан, панобиностат) и их комбинации, согласно принятым схемам терапии [2]. В литературе также описаны такие виды лечения, как трансплантация собственных стволовых клеток, радиотерапия и оперативное лечение (направлено на устранение сдавления спинного мозга при компрессионных переломах позвонков), что в некоторых случаях позволяет снижать частоту возникновения и тяжесть побочных эффектов заболевания и контролировать связанные c ММ осложнения [16–18].
Клинический случай
Пациент М. 61 года. Поступил в гастроэнтерологическое отделение клиники Петра Великого СЗГМУ им. Мечникова (Санкт-Петербург) 01.02.2017 для уточнения причин гепатомегалии и резкого снижения эластичности печени (эластичность печени по результатам фиброэластометрии соответствовала стадии фиброза F4). При поступлении предъявлял жалобы на общую слабость, чувство дискомфорта и сдавления в правом подреберье, боли в поясничном отделе позвоночника.
Краткий анамнез: в сентябре 2014 г. прооперирован по поводу меланомы левой пяточной области T1M0N0, произведено иссечение образования. Гистологическое заключение от 29.09.2014: эпителиоидно-клеточная меланома кожи левой пяточной области, не изъязвляющая эпидермис, с умеренной инфильтрацией и малым количеством пигмента. Уровень инвазии по Кларку II, толщина по Бреслоу – 0,9 мм.
В мае 2016 г. в биохимическом анализе крови выявлено повышение активности ЩФ до 300 ЕД/л (норма – до 270 ЕД/л). При мультиспиральной компьютерной томографии (МСКТ) брюшной полости от апреля и июля 2016 г. описано патологическое образование левого надпочечника, других изменений не выявлено. В июле 2016 г. возникли интенсивные боли в правом подреберье, вызвал скорую медицинскую помощь, госпитализирован в клинику Военно-медицинской академии. При ультразвуковом исследовании (УЗИ) выявлены конкременты желчного пузыря, проведен курс спазмолитической терапии. Выписан с диагнозом «желчнокаменная болезнь», рекомендована плановая холецистэктомия. После выписки пациента несколько раз в неделю беспокоили боли в правом подреберье, появились желтушность кожных покровов, кожный зуд, обесцвечивание кала. При амбулаторном обследовании выявлено повышение активности ЩФ до 4,8 нормы (1300 ЕД/л), ГГТП до 8,8 нормы (618 ЕД/л). Пациенту рекомендован прием урсодезоксихолевой кислоты (УДХК), адеметионина. На фоне терапии отмечал улучшение самочувствия, болевой абдоминальный синдром не беспокоил, цвет кожных покровов и окраска кала нормализовались.
В сентябре 2016 г. выполнена очередная МСКТ, выявлено значительное увеличение размеров печени до 20 см.
Пациенту было проведено расширенное обследование:
- при серологическом исследовании выявлен пограничный уровень антинуклеарных антител (ANA hep-2) – 1:160, мелкогранулярный тип свечения (норма менее 1:160);
- при сцинциграфии с 99Tc выявлены сцинциграфические признаки патологических изменений передних отрезков 2-го, 4, 5, 6, 7-го ребер справа, передних отрезков 4-го, 5-го ребер слева, диффузное накопление радиофармакологического препарата в печени, что является косвенным признаком диффузного заболевания печени;
- в декабре 2016 г. проведено стацио-нарное обследование в онкологическом отделении для исключения рецидива меланомы. УЗИ органов брюшной полости от 03.12.2016: эхо-признаки выраженной гепатомегалии, диффузных изменений паренхимы печени, изменения кавального кровотока (косвенные признаки повышения жесткости), начального расширения внутрипеченочных протоков в левой доле, холестероза желчного пузыря. Рекомендовано выполнение исследования для оценки выраженности фиброза печени;
- 06.12.2016 пациенту выполнена эластометрия печени, выявлены признаки фиброза степени – F4 (75 kPa), степень стеатоза – S0 (182 dB/m). Надо признать, что в ряде случаев данные о выраженном фиброзе/циррозе печени, полученные при неинвазивных методах исследования, которые получили широкое распространение в клинической практике в последнее время, мотивируют и врача, и пациента на более глубокое обследование;
- при обследовании на онкомаркеры: повышение уровня CA-19-9 до 74,3 (норма до 35). Рекомендована госпитализация с целью установления этиологии изменений в печени и определения дальнейшей тактики ведения.
При обследовании в отделении гастроэнтерологии в феврале 2017 г. по данным эзофагогастродуоденоскопии признаков портальной гастропатии варикозного расширения вен пищевода не выявлено. УЗИ органов брюшной полости: признаки гепатомегалии (Косой вертикальный размер правой доли – 230 мм, толщина левой доли – 154 мм), начальные проявления портальной гипертензии (диаметр воротной вены – 13 мм). Холедох не расширен (5 мм). Мелкие конкременты желчного пузыря, сгущение желчи. Размеры селезенки: продольный – 107 мм, переднезадний – 35 мм, диаметр селезеночной вены – 8 мм.
В биохимическом анализе крови от 27.02.2017 обращали на себя внимание следующие изменения: незначительное повышение уровня общего белка (87 г/л при норме до 85 г/л) при нормальном уровне альбумина крови, повышение активности аспартатаминотрансферазы (АСТ) до 1,5 нормы, повышение уровня ЩФ до 7 норм, уровень общего билирубина – 20,3 мкмоль/л (норма – до 19 мкмоль/л), холестерин – 7,9 ммоль/л (норма – до 5,2 ммоль/л), а также повышение уровня креатинина до 202 мкмоль/л (при норме до 115 мкмоль/л), мочевины – до 13,2 ммоль/л (при норме до 8,3 ммоль/л), выраженное снижение скорости клубочковой фильтрации (31 мл/мин/1,73 м2). При повторном исследовании ANA hep-2 титр менее 1:40. В клиническом анализе крови выявлена гипохромная анемия легкой степени (гемоглобин=113 г/л, при норме от 130 г/л), незначительный лейкоцитоз (9,4×109/л, при норме до 9,0×109/л), без отклонений формулы, уровень тромбоцитов в пределах референсных значений. В коагулограмме незначительное увеличение активированного частичного тромбопластинового времени, протромбинового времени, международного нормализованного отношения в пределах референсных значений. При исследовании иммуноглобулинов в сыворотке крови выявлено клинически значимое повышение уровней IgG, уровни IgA и IgМ не повышены. Исследование сыворотки крови на легкие цепи k и λ-типа от 27.02.2017: значимое повышение легких цепей иммуноглобулинов k- и λ-типа.
Компьютерная томография брюшной полости была выполнена пациенту 10.02.2017: выявлена гепатомегалия (вертикальный размер правой доли печени 240 мм), контуры ее четкие, ровные, структура паренхимы однородная. Снижение плотности печени до 43–53 HU. Также выявлен компрессионный перелом тела L1 cо снижением его высоты на 1/2 и смещением верхнего отломка кзади на 4 мм с сужением позвоночного канала (ширина позвоночного канала на уровне Th12 составляет 14 мм, на уровне диска Th12-L1 – 10 мм, на уровне тела L2 – 14 мм).
Для оценки вероятности фиброза/цирроза печени рассчитан индекс APRI (AST to Platelet Ratio Index) по формуле: АСТ/верхний предел АСТ×100/тромбоциты (109/л). APRI=0,43, что свидетельствует в пользу отсутствия у пациента значимого фиброза [19]. Следует указать, что полученные данные полностью противоречили результатам, полученным при фиброэластометрии в 2016 г., что лишний раз указывает на необходимость комплексного подхода при оценке фиброза и стеатоза печени с помощью неинвазивных методов исследования [20, 21].
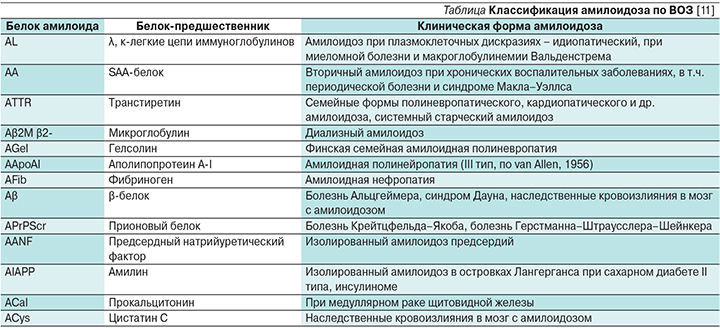
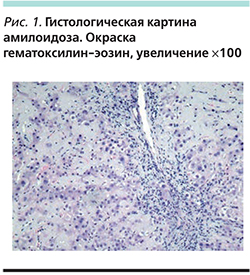 Для уточнения диагноза и возможной причины поражения печени пациенту была выполнена биопсия печени. По данным гистологического заключения портальные тракты умеренно расширены за счет разрастания соединительной ткани с формированием септ, очаговой лимфоцитарной инфильтраци без инвазии в дольки; печеночные балки резко деформированы за счет интралобулярного отложения гомогенных эозинофильных масс, слабо окрашиваемых конго-красным. Стадия фиброза – F2–3 (по шкале METAVIR). Гистологическая картина больше соответствует амилоидозу (рис. 1–4). При иммуногистохимическом исследовании биоптата выявлен AL/λ-амилоидоз, нельзя исключить наличие множественной миеломы.
Для уточнения диагноза и возможной причины поражения печени пациенту была выполнена биопсия печени. По данным гистологического заключения портальные тракты умеренно расширены за счет разрастания соединительной ткани с формированием септ, очаговой лимфоцитарной инфильтраци без инвазии в дольки; печеночные балки резко деформированы за счет интралобулярного отложения гомогенных эозинофильных масс, слабо окрашиваемых конго-красным. Стадия фиброза – F2–3 (по шкале METAVIR). Гистологическая картина больше соответствует амилоидозу (рис. 1–4). При иммуногистохимическом исследовании биоптата выявлен AL/λ-амилоидоз, нельзя исключить наличие множественной миеломы.
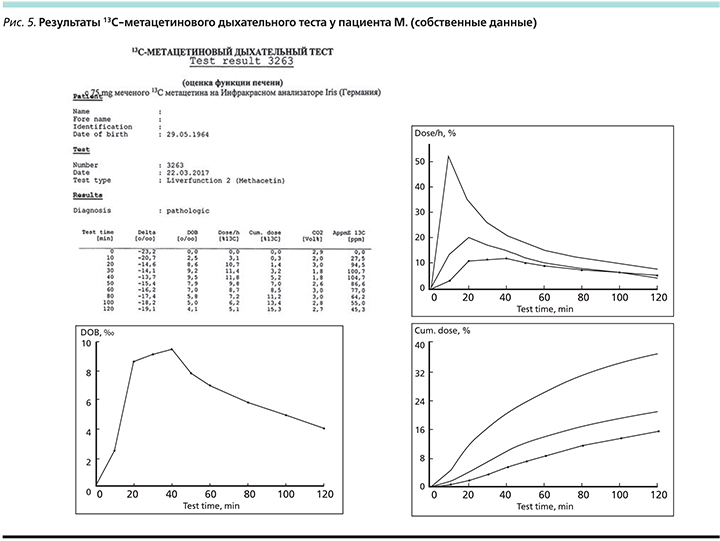
Для оценки функциональной активности печени пациенту было выполнено исследование – 13С-метацетиновый дыхательный тест (рис. 5): выявлено значительное снижение функционального резерва, а также снижение скорости и объема метаболизма в системе цитохрома Р4501А2, что свидетельствует о длительном поражении печени.
На основании проведенных обследований пациенту был установлен диагноз: множественная миелома IgG/каппа IIB-стадии по Дьюри–Салмону, III стадии по ISS, остеодеструктивный синдром. Вторичный AL-амилоидоз с поражением печени (F4 по данным эластометрии, дисфункция печени тяжелой степени по данным 13С-МДТ). Осложнения: хроническая болезнь почек 4, хроническая сердечная недостаточность IIa. портальная гипертензия. Сопутствующий диагноз: желчнокаменная болезнь, хронический калькулезный холецистит.

Пациент был направлен на лечение к гематологу. В настоящее время проводятся курсы химиотерапии, на фоне чего купированы боль в правом подреберье и желтуха, уменьшился астенический синдром. За время наблюдения гематологом имеется тенденция к уменьшению размеров печени, однако прогноз пациента остается неблагоприятным.
Данный клинический пример иллюстрирует клинические особенности течения множественной миеломы, осложненной вторичным системным AL-амилоидозом: бессимптомное начало заболевания, стремительное увеличение размеров печени с формированием признаков холестаза, портальной гипертензии, изменения в структуре костей (компрессионный перелом L1), потребовавшего проведения дифференциального диагноза с синдромосходными заболеваниями.
Заключение
Целью данной статьи стало привлечение внимания врачей к трудностям дифференциальной диагностики ММ. Актуальность проблемы диагностики, дифференциальной диагностики и эффективного лечения по-прежнему высока. Данное заболевание протекает практически бессимптомно, диагностические критерии неоднозначны. Диагностика занимает длительное время, зачастую первыми выявляются осложнения данного заболевания, такие как системный AL-амилоидоз, часто требующий проведения дифференциального диагноза с синдромосходными заболеваниями.
Следует обратить внимание практических врачей на осторожность и аккуратность при интерпретации данных по оценке фиброза печени, полученных при выполнении неинвазивных методов исследования (фиброэластометрия печени, фибротесты, онлайн-калькуляторы и др.) и прежде всего у полиморбидных пациентов. При наличии результатов исследований, полученных с помощью нескольких неинвазивных методов, противоречащих друг другу или значимо различающихся, целесообразно рекомендовать проведение пункционной биопсии печени.


