
Основным источником образования аммиака в организме человека является азот пищевого белка, образующийся в ходе реакций дезаминирования аминокислот в печени. Дополнительными источниками образования аммиака являются:
- Разложение мочевины и белка уреазаположительной микрофлорой желудочно-кишечного тракта (ЖКТ).
- Образование аммиака в мышечной ткани при физической нагрузке.
- Распад глутамина в тонкой кишке.
- Абсорбция аммиака в почках при гипокалиемии и/или алкалозе.
Выделение мочевины осуществляется преимущественно через почки (около 80%), примерно 20% мочевины повторно поступают в ЖКТ, где вновь разлагается уреазаположительными бактериями до аммиака.
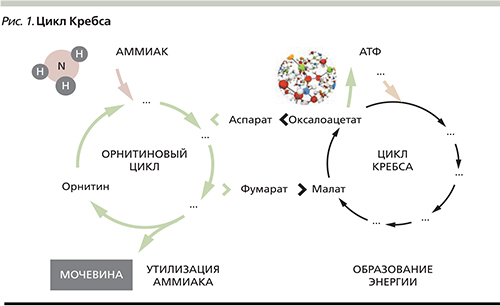
Детоксикация аммиака в организме осуществляется преимущественно в митохондриях перипортальных гепатоцитов за счет связывания в орнитиновом цикле с аминокислотами и образованием нетоксичной мочевины (рис. 1). Частично детоксикация аммиака происходит в мышечной ткани в процессе синтеза глутамина при участии фермента глутаминсинтетазы. Эта реакция с меньшей интенсивностью протекает также в астроцитах головного мозга и перивенозных гепатоцитах печени. Образующийся в результате этих превращений глутамин нетоксичен и выделяется с мочой.
Таким образом, являясь основным источником аммиака, печень в то же время служит главным местом его обезвреживания. Именно поэтому гипераммониемия развивается в организме человека, прежде всего при хронических заболеваниях печени (ХЗП). К причинам этого относятся снижение активности орнитинового цикла и глутаминсинтетазной реакции при печеночно-клеточной недостаточности и порто-системное шунтирование при развитии и прогрессировании портальной гипертензии.
Значительно реже в рутинной клинической практике встречаются генетически детерминированные ферментопатии, которые сопровождаются повышением концентрации аммиака в сыворотке крови. В зависимости от дефицита или дефекта того или иного фермента выделяют несколько видов генетических заболеваний: гипераммониемию типа I (в основе – дефект карбамоилфосфатсинтетазы I), гипераммониемию типа II (в основе – дефект орнитинкарбамоилтрансферазы), цитруллинемию (в основе – дефект аргининосукцинатсинтетазы), аргининосукцинатурию (в основе – дефект аргининосукцинатлиазы), гипераргининемию (в основе – дефицит аргиназы).
Известно, что аммиак является одним из важнейших нейротоксических метаболитов в организме человека. В клинической практике наиболее частым проявлением гипераммониемии является печеночная энцефалопатия (ПЭ) при патологии печени, представляющая собой спектр нервно-психических расстройств на фоне острой или хронической печеночно-клеточной недостаточности и/или портосистемном шунтировании крови. В основе патогенеза ПЭ лежит дисбаланс аминокислот в головном мозге, приводящий к отеку астроглии и нарушениям ее функций, таких как изменения постсинаптических рецепторов и процессов нейротрансмиссии, нарушение проницаемости гематоэнцефалического барьера, снижение энергетического обеспечения нейронов. Степень клинических проявлений напрямую коррелирует с уровнем аммиака в сыворотке крови. В зависимости от выраженности нарушений деятельности головного мозга выделяют четыре степени тяжести печеночной энцефалопатии (от минимальной до комы).
В последние годы в связи с совершенствованием диагностических методик актуально отделение клинически выраженных стадий ПЭ (дезориентация, атаксия, кома) от стадии с минимально выраженными проявлениями (латентная ПЭ). Такую ПЭ можно выявить, используя специальные опросники или метод вызванных потенциалов головного мозга. При этом выявляются когнитивные и психомоторные расстройства, такие как трудности с принятием решений, снижение скорости психомоторных реакций и др. К клиническим симптомам латентной ПЭ относятся повышенная утомляемость, слабость, раздражительность, инверсия сна (сонливость днем и бессонница ночью), нарушения речи, изменения почерка, рассеянность за рулем и при выполнении работы, требующей повышенной концентрации внимания, тремор, снижение мышечных рефлексов.
К сожалению, результатом таких нарушений могут стать серьезные дорожно-транспортные происшествия с тяжелыми последствиями [1, 2]. В связи с этим выявление минимальной ПЭ имеет большое значение для работников многих профессий: водителей автотранспорта, операторов на автоматизированном оборудовании и др.
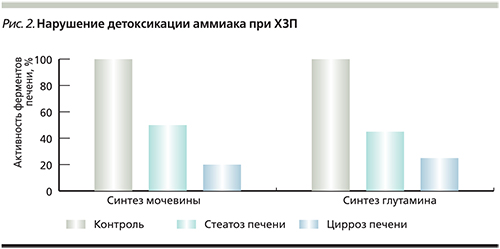
В исследованиях показано, что у больных стеатозом печени при отсутствии клинических признаков воспаления и печеночно-клеточной недостаточности уже имеется выраженное снижение детоксикации аммиака по обоим путям (уменьшается синтез и мочевины, и глутамина) за счет выраженного снижения активности соответствующих ферментов в печени (рис. 2).
В 2016 г. ученые из Великобритании получили новые научные данные, свидетельствующие о том, что гипераммониемия активирует звездчатые клетки печени (ЗКП) и как следствие – может приводить к усиленному коллагенообразованию и прогрессирующему фиброзированию [3].
Известно, что ЗКП являются основными профиброгенными клетками органа. Хроническое повреждение печени под влиянием различных этиологических факторов (алкоголь, вирусная инфекция, лекарства, холестаз и др.) способствует их активации и дифференцировке в миофибробластоподобные клетки, которые приобретают сократительные, провоспалительные и фиброгенетические свойства. При этом ЗКП пролиферируют, из них исчезают капли жира, увеличивается шероховатая эндоплазматическая сеть, в них появляется специфический белок гладких мышц (α-актин), увеличивается количество рецепторов к цитокинам, стимулирующим пролиферацию и фиброгенез.
К факторам, активирующим ЗКП, относятся трансформирующий фактор роста (TGF-β1 – Transforming growth factor beta), тромбоцитарный фактор роста (PDGF – Platelet-derived growth factor), фактор роста фибробластов, интерлейкин-1 (ИЛ-1), эпидермальный фактор роста, фактор некроза опухоли α. Среди всех факторов роста TGF-β1 позиционируется как ключевой медиатор в фиброгенезе у человека. Различные способы, воздействующие на синтез этого фактора или на сигнальные пути, которые реализуются с участием этого фактора, значимо снижают фиброз в экспериментальных моделях [4].
Активированные ЗКП мигрируют и аккумулируются в месте поражения ткани печени, при этом секретируя большое количество внеклеточного матрикса и одновременно регулируя деградацию этих молекул на уровнях транскрипции и посттранскрипции. Повышение содержания информационной коллагеновой РНК является опосредующим фактором, повышающим синтез коллагена активированными ЗКП. В этих клетках посттранскрипционная регуляция коллагена осуществляется путем последовательности 3-го нетранслируемого региона РНК-связующего протеина aСР2, равно как и посредством структуры в 5-м окончании коллагена информационной РНК. Кроме этого ЗКП экспрессируют большое количество нейроэндокринных маркеров (реелин, нестин, нейротрофины, синаптофизин и глиально-фибриллярные кислотные протеины), а также несут рецепторы нейротрансмиттеров, выделяют провоспалительные цитокины, нейрофильный и моноцитарный хемоаттрактаны, которые усиливают воспалительную реакцию в пораженной печени [5].
Фиброз печени является основным, этиологически независимым путем прогрессирования хронических диффузных заболеваний печени вплоть до цирроза. Печеночный фиброз ассоциируется с изменением количества и качественного состава экстрацеллюлярного коллагенового матрикса (ЭКМ). При выраженных стадиях фиброза печень содержит приблизительно в 6 раз больше ЭКМ, чем в норме, а в его составе определяются коллагены (1-го, 3 и 4-го типов), фибронектин, ундулин, эластин, ламинин, гиалуронан и протеогликаны. Снижение скорости резорбции ЭКМ и выведение молекул металлопротеиназ являются в основном следствием перевысвобождения их специфических ингибиторов (TIMPs – tissue inhibitors of metalloproteinases). Результатом превалирования процессов образования внеклеточного матрикса над его разрушением является формирование фиброзного рубца, при этом фиброз на ранних стадиях развития – процесс обратимый, а цирроз с характерными сшивками между коллагеновыми волокнами и узлами регенерации необратим. Прогрессирующее накопление и отложение внеклеточного матрикса в пространстве Диссе приводят к исчезновению фенестров эндотелия, капилляризации и стенозированию синусоидов с постепенным развитием портальной гипертензии [6–8].
Таким образом, в прогрессировании ХЗП главенствующую роль играют повреждения и ишемия гепатоцитов, которые запускают регенераторные процессы воспаления и коллагенообразования. В свою очередь повышенное коллагенообразование из-за избыточного отложения внеклеточного матрикса и нарушения портального кровотока также приводит к ишемии и некрозу гепатоцитов. Таким образом, можно говорить о «circulus vitiosus» (порочном круге) в прогрессировании заболеваний печени.
Почему ученые из Великобритании считают, что гипераммониемия способна выступить важным фактором в этом «circulus vitiosus»? Такие выводы были сделаны на основе результатов собственного исследования, состоявшего из двух частей: in vitro и in vivo. Первичные ЗКП, полученные из печени здоровых доноров (hHSCs – human hematopoietic stem cells), были посеяны (плотность – 26×103/см2) в исходных, богатых сывороткой условиях (CM – полная среда) на 24 часа с последующим удалением сыворотки на следующие 24 часа (SFM – Serum-free medium). Экзогенный глутамин был удален из культуральной среды во избежание искажения результатов эксперимента.
In vitro показано, что аммиак дозозависимо снижает клеточную пролиферацию и метаболизм в первичных человеческих ЗКП, но не вызывает гибели клеток.
Длительная обработка клеток аммиаком (до 72 часов) индуцирует развитие и прогрессирование эндоретикулярного стресса в hHSCs, что проявлялось выраженным перинуклеарным накоплением красителя (ER-Tracker™ Red) и появлением цитоплазматических вакуолей по мере роста концентрации аммиака. Использование Image IT™-набора для определения зеленых активных форм кислорода позволило исследователям выявить аммиак-дозозависимое образование активных радикалов кислорода (ROS – Reactive oxygen species) в hHSCs. Таким образом, установлено, что увеличение концентрации аммиака и время его воздействия напрямую влияют на уровни мРНК экспрессии маркеров стресса [3].
Исследование также показало, что при повышенных концентрациях аммиака hHSCs приобретают выраженный профиброгенный и провоспалительный потенциал, что подтверждается результатами исследования in vitro:
- аммиак значительно увеличивает экспрессию белка α-SMA, являющегося активатором ЗКП, а при уровне аммиака 300 мкМ увеличивается синтез виментина – важного промежуточного филамента;
- возрастание концентраций аммиака сопровождается увеличением уровней миозина IIa (играющего ключевую роль в сокращении HSC), миозина IIb (важный фактор активации HSC); экспрессией p38 MAPK, ростом уровней PDGF-Rβ и коллагена I типа;
- повышение уровней аммиака индуцирует сильный и значимый рост мРНК экспрессии металлопротеиназ-2, в то время как мРНК-экспрессия TIMP1 снижается;
- при обработке hHSC аммиаком в концентрации 300 мкМ на протяжении 72 часов мРНК значительно возрастает экспрессия провоспалительного ИЛ-1β;
- аммиак в дозах 50 и 100 мкМ значительно увеличивает в hHSC мРНК- экспрессию ИЛ-6, но не изменяет мРНК-экспрессию ИЛ-8.
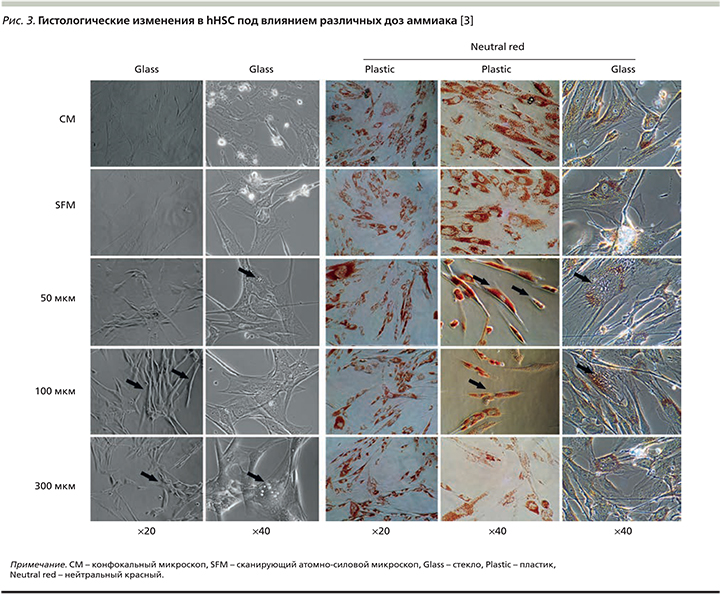
При гистологическом изучении клеток установлено, что аммиак дозозависимо вызывает серьезные морфологические изменения. Так, при световой микроскопии и в тесте жизнеспособности с нейтральным красным (20х, 40х) наблюдали превращение миофибробластоподобных клеток в веретеноподобные фибробласты под действием повышенных концентраций аммиака (рис. 3). Электронная микроскопия показала изменения структуры цитоскелета клеток с образованием цитоплазматических вакуолей при гипераммониемии, однако эти изменения быстро регрессировали после помещения клеток в безаммиачную среду [3].
Вторая часть исследования была проведена на самцах крыс, которые были разделены на три группы: в двух группах проведено перевязывание общего желчного протока (BDL – экспериментальная модель холестаза) с целью формирования экспериментального повреждения печени, при этом крысам одной из групп вводился орнитин (ОР), второй – физиологический раствор. Третья группа животных была контрольной, без повреждения печени.
Анализ результатов опыта показал, что концентрации аммиака были значительно увеличены в плазме крыс с BDL по сравнению с контролем (182±12,8 против 62,51±6,2 мкМ; p<0,0001), при лечении OP уровни аммиака значительно снижались (83,8±16,3 мкМ; p<0,0001). Изменения уровней аммиака в плазме прямо коррелировали с изменениями портального давления, которое было значительно выше у крыс с BDL по сравнению с крысами контроля (14±0,6 против 5,5±0,3 мм рт.ст.; p<0,0001). Снижение уровня аммиака на фоне лечения OP сопровождалось значительным снижением портального давления по сравнению с крысами с BDL, получавшими физиологический раствор (11,3±0,3 мм рт.ст.; p<0,01).
Дополнительным фактором риска портальной гипертензии при гипераммониемии является нитрозативный стресс, являющийся причиной нарушений печеночной гемодинамики. Так, у крыс с BDL наблюдали статистически значимое по сравнению с контрольной группой (p<0,001) увеличение белковой экспрессии показателей возросшего нитрозативного стресса: индуцированной NO синтазы (iNOS), эндотелиальной NO синтазы (eNOS), кавеолина-1 (внутриклеточного ингибитора eNOS) и 3-N тирозина. Прием OP значительно (p<0,01) снижал экспрессию iNOS, кавеолина-1 и 3-N тирозина (В и С). Сниженная ферментативная активность eNOS у крыс с BDL, усилившая нитрозативный стресс, также была восстановлена после лечения OP до практически нормальных уровней (p<0,01).
Таким образом, in vitro и in vivo исследовано многообразие влияний аммиака на функциональную активность печени [3]. Показано, что гипераммониемия является причиной следующих нарушений:
- активации ЗКП,
- снижения клеточного метаболизма и пролиферации ЗКП,
- активации профиброгенного/провоспалительного профиля ЗКП,
- нарушения внутрипеченочной гемодинамики и нарастания портального давления,
- стимуляции эндоретикулярного стресса,
- индукции образования активных форм кислорода.
Таким образом, в список факторов, активирующих ЗКП и имеющих большое значение в прогрессировании ХЗП, необходимо внести гипераммониемию. В связи с этим с целью снижения уровней аммиака чрезвычайно актуальным представляется применение L-орнитин-L-аспартата (LOLA, Гепа-Мерц), препарата, обеспечивающего обезвреживание аммиака в клетках печени, мышцах и астроцитах головного мозга. Важной особенностью L-орнитин-L-аспартата является непосредственное участие препарата в орнитиновом цикле обезвреживания аммиака.
В многочисленных исследованиях доказана гипоаммониемическая эффективность LOLA, позволяющая достоверно снижать проявления ПЭ. Так, лечение минимальной ПЭ у больных циррозом печени сопровождается достоверным снижением уровней аммиака в сыворотке крови, коррелирующим со снижением выраженности ПЭ, снижением частоты нарушений Правил дорожного движения с 0,82 до 0,51 и снижением частоты дорожно-транспортных происшествий с 0,18 до 0,0039 [1].
Гепатопротективные свойства LOLA показаны в отношении пациентов с ХЗП различной этиологии [9,10,11]. Данные многоцентрового нерандомизированного проспективного когортного исследования, проведенного в Германии, с участием 1167 пациентов с ХЗП, в т.ч. 648 больных неалкогольным стеатогепатитом, продемонстрировали эффективность и хорошую переносимость LOLA [12].
Нарастание концентрации аммиака выявляется у больных ХЗП уже на доцирротической стадии [9]. В исследовании с участием больных неалкогольной жировой болезнью печени показано, что на фоне применения L-орнитин-L-аспартата гипераммониемия, исходно имевшаяся на 0–1-й стадии фиброза, существенно снизилась, что сопровождалось улучшением общего состояния и лабораторных показателей. В другом российском исследовании с использованием реогепатографии было убедительно показано нарушение портального кровотока при гипераммониемии у пациентов с доцирротическими стадиями ХЗП [13]. Лечение 289 пациентов с неалкогольным стеатогепатитом с использованием LOLA на протяжении трех месяцев на фоне хорошей переносимости и высокой приверженности больных способствовало снижению уровней аммиака, коррелирующему с уменьшением сосудистых нарушений, статистически значимому улучшению клинико-биохимических показателей и качества жизни [14].
В декабре 2015 г. МЗ РФ в инструкцию препарата L-орнитин-L-аспартат были внесены важные изменения. Теперь показаниями к применению L-орнитин-L-аспартат кроме ХЗП, сопровождающихся ПЭ, являются стеатозы и стеатогепатиты различного генеза. В ноябре 2016 г. L-орнитин-L-аспартат внесен в рекомендации Научного общества гастроэнтерологов России и Российского научного медицинского общества терапевтов по диагностике и лечению неалкогольной жировой болезни печени (2-я версия).
Таким образом, снижение уровня аммиака является патогенетическим подходом к лечению ХЗП, позволяющим не только снижать проявления ПЭ, но и уменьшать активацию ЗКП, улучшать печеночный кровоток и препятствовать развитию и прогрессированию фиброза печени.


