
Введение
Принято считать, что диагностика и лечение тромбоцитопении (ТП) находятся в сфере профессиональной компетенции специалиста-гематолога. Вместе с тем к настоящему моменту накоплена масса данных, свидетельствующих о снижении числа тромбоцитов при различных патологических состояниях. Это объясняет необходимость более широкого обсуждения данной проблематики среди врачей различных клинических специальностей.
Общие положения
Термин «тромбоцитопения» обычно используют при уровне тромбоцитов ниже 100,0×109/л, хотя нормальным уровнем тромбоцитов принято считать таковой в пределах от 150,0×109/л до 450,0×109/л [1]. Поэтому ряд экспертов выделяют латентную ТП при уровне тромбоцитов от 100,0×109/л до 150,0×109/л [2]. Выделение латентной ТП, на наш взгляд, обоснованно с практической точки зрения. С одной стороны, такая клиническая ситуация требует динамического наблюдения за уровнем тромбоцитов независимо от причины, с другой – при числе тромбоцитов 100,0×109/л и более полностью обеспечивается гемостаз, что, как указывается в большинстве имеющихся руководств, безопасно в контексте риска развития кровотечений [3] и что позволяет проводить различные оперативные вмешательства, в т.ч. и родоразрешение, при указанном уровне тромбоцитов.
Более того, концентрация тромбоцитов от 100×109/л до 50×109/л, протекающая без спонтанного геморрагического синдрома, может также считаться безопасной. В случаях появления признаков кровоточивости при указанном числе тромбоцитов следует искать дополнительный фактор, провоцирующий геморрагический синдром, или учитывать наличие сосудистой патологии, например, у пациентов преклонного возраста. Существующие подходы указывают, что коррекцию ТП следует проводить при числе тромбоцитов от 50×109/л до 30×109/л только при наличии геморрагических проявлений. Критическим для развития опасных для жизни геморрагических проявлений является содержание тромбоцитов ниже 10,0×109/л. Пациенты с таким уровнем ТП нуждаются в безотлагательной терапии независимо от степени клинических проявлений геморрагического синдрома [3, 4].
Этиопатогенез ТП
Говоря о ТП, следует подчеркнуть разнообразие приводящих к ней патогенетических механизмов и еще большее количество патологических состояний, при которых данные патогенетические механизмы реализуются [5]. Выделяют два основных механизма патогенеза ТП (табл. 1): нарушение образования тромбоцитов и повышенное их разрушение.
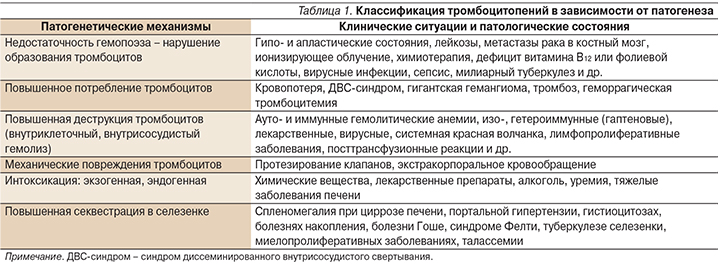
Диагностические алгоритмы при ТП
При выявлении у пациента ТП весьма важен детальный анализ истории заболевания, в частности установление предшествовавших развитию ТП факторов: бактериальная или вирусная инфекция, в т.ч. и вирусные гепатиты; вакцинация или применение лекарственных препаратов; пребывание в странах с риском заражения инфекционными заболеваниями (малярия, риккетсиоз, лихорадка Денге и др.); употребление алкогольных и хинин-содержащих напитков; варикозная болезнь, тромбозы, сердечно-сосудистая патология и ее терапия антикоагулянтами и дезагрегантами; наличие сопутствующих заболеваний, особенно аутоиммунных, инфекционных или опухолевых, протекающих с ТП, ДВС-синдромом; трансфузии; пересадка органов в анамнезе; беременность; наличие и длительность кровотечений после хирургических вмешательств.
В семейном анамнезе обязательно уточнить наличие болезни системы кроветворения у родственников.
При проведении объективного исследования следует активно выявлять такие симптомы, как гипо- или гипертермия, снижение массы тела, симптомы интоксикации, гепато- и спленомегалия, лимфаденопатия, патология молочных желез, сердца, вен нижних конечностей, а также врожденные аномалии. Все эти данные не специфичны.
Лабораторно-инструментальная диагностика включает несколько направлений. В клиническом анализе крови обязательны оптический подсчет числа тромбоцитов и оценка морфологии тромбоцитов (микроформы и гигантские тромбоциты), при этом следует помнить, что при наличии агрегатов тромбоцитов для исключения «ложной» ТП при использовании консерванта ЭДТА (этилендиаминтетраацетат кальция-натрия) необходим повторный анализ (используется пробирка с цитратом натрия). Всем пациентам проводится ультразвуковое исследование или компьютерная томография (КТ) органов брюшной полости и забрюшинного пространства, рентгенография или КТ органов грудной клетки, а также обследование для исключения онкологических заболеваний. Принимая во внимание, что идиопатическая тромбоцитопеническая пурпура (ИТП) – диагноз исключения, спектр используемых лабораторных тестов достаточно широк (табл. 2), по своей диагностической значимости их подразделяют на обязательные, потенциально полезные и тесты с недоказанной информативностью.

Принципы лечения ИТП
В лечении ИТП выделяют несколько линий терапии. На первом этапе используются гормоны (преднизолон, дексаметазон) и специфический иммуноглобулин. К терапии второй линии относят спленэктомию и стимуляторы тромбопоэза (миметики тромбопоэтина). Ритуксимаб и другие препараты с иммуносупрессивным действием (азатиоприн, циклофосфамид, циклоспорин А, винкристин, винбластин) рассматриваются как средство резерва (третья линия терапии) [4].
ТП в гастроэнтерологии
Принимая во внимание множественность причин ТП, необходимо проводить исключение заболеваний, которые могут в клинической картине иметь снижение уровня тромбоцитов; в практике врача-гастроэнтеролога это прежде всего H. pylori-ассоциированные заболевания и поражения печени различного генеза.
В 1998 г. А. Gasbarrini и соавт. опубликовали в журнале «Ланцет» статью «Regression of autoimmune thrombocytopenia after eradication of Helicobacter pylori», положившую начало новому направлению в изучении хеликобактериоза, сопутствующих с ним состояний, а также более тесному взаимодействию врачей двух клинических специальностей, а именно гатроэнтерологов и гематологов [6]. Ранее было показано, что аутоиммунные реакции посредством перекрестной мимикрии между антигенами системы Lewis эпителиальных клеток желудка и H. pylori могут играть определенную роль в хеликобактер-индуцированном повреждении слизистой оболочки; моноклональные анти-H. pylori-антитела вступают в реакции взаимодействия с тканями вне желудочно-кишечного тракта, а именно с эпителиоцитами протоков слюнных желез и почечных канальцев [7]; поликлональные анти-H. pylori-антитела взаимодействуют с капиллярами почечных клубочков, что может приводить к мембранозной нефропатии [8]. Опираясь на данные предшествовавших исследований, в частности на результаты наблюдений [9] о купировании проявлений болезни Шенлейн–Геноха после успешно проведенной эрадикационной терапии, А. Gasbarrini и соавт. предположили патофизиологическую связь между ИТП и хронической инфекцией H. pylori. Они описали повышение уровня тромбоцитов у всех 8 пациентов ИТП, получивших эрадикационную терапию, при этом уровень тромбоцитов у 3 человек, такой терапии не получавших, оставался неизменным.
К настоящему времени опубликована масса работ, систематических обзоров, исследовавших взаимосвязь ИТП и H. pylori, механизмы развития данной взаимосвязи, выявляющих предикторы положительного ответа на эрадикационную терапию.
Предложено несколько гипотез механизмов, с помощью которых H. pylori вызывает развитие ИТП. Считается, что образуются перекрестно-реактивные антитела, взаимодействующие как с компонентами H. pylori, так и с поверхностными антигенами тромбоцитов посредством молекулярной мимикрии. Итальянские ученые во главе с R. Scandellari показали, что заражение штаммами H. pylori, экспрессирующими CagA (cytotoxin-associated gene A), может быть причиной хронических случаев ИТП. По данным проведенного ими исследования, увеличение числа тромбоцитов после эрадикационной терапии наблюдалось у 43% пациентов после 6 месяцев наблюдения. Пациенты были сопоставимыми по всем основным клиническим признакам, за исключением одного: сывороточные антитела к CagA присутствовали у 83% пациентов, ответивших на терапию, и только у 12,5% не ответивших (p=0,026). Авторы пришли к выводу, что антибиотикотерапия H. pylori-инфекции может приводить к исчезновению иммунных перекрестно реагирующих антител и, следовательно, может рассматриваться как путь терапии пациентов с ИТП, особенно для тех лиц, у которых выявляются антитела к CagA [10]. Впоследствии, опираясь на данные, полученные R. Scandellari и соавт., а также принимая во внимание тот факт, что почти все CagA-положительные штаммы H. pylori также экспрессируют вакуолизирующий токсин, группа исследователей во главе с N. Figura предположили существование молекулярной мимикрии некоторых тромбоцитарных пептидов с вакуолизирующим цитотоксином А H. pylori. В частности, компонент тромбоцитов, связывающий домен рецептора 289 аминокислот для фактора фон Виллебранда (GP1b-тромбоцит), показал определенное структурное сходство с VacA- H. pylori [11]. M. Michel и соавт. [12] также исследовали гипотезу молекулярной мимикрии и обнаружили, что тромбоциты инфицированных H. pylori-пациентов ИТП, обладающие способностью реагировать с GPIIb/IIIa или GPIb, не могут распознавать антигены H. pylori. С другой стороны, T. Takahashi и соавт. [13] сообщили, что тромбоциты пациентов с ИТП, инфицированных H. pylori, «распознавали» CagA при реакции иммуноблоттинга, а тромбоциты пациентов, инфицированных H. pylori, но без ИТП – нет. Y. Bai и соавт. изучили перекрестное взаимодействие моноклонального антитела против уреазы В хеликобактера с поверхностным гликопротеином тромбоцитов GPIIb/IIIa и предположили, что иммунный ответ на UreB может участвовать в патогенезе ИТП [14]. Все эти данные свидетельствуют о том, что перекрестно реагирующие антитела против H. pylori могут присутствовать у пациентов с ИТП, но их патогенетическая роль до конца не ясна.
В качестве еще одного механизма рассматривается тот факт, что хроническая инфекция H. pylori может неспецифически воздействовать на иммунную систему хозяина, стимулируя приобретенные иммунные ответы через выработку Т- и В-аутоантител. Японские исследователи S. Yamanishi и соавт. [15] показали, что уреаза H. pylori способна инициировать аутоиммунные реакции путем активации В1-клеток, однако это не объясняет развития специфичного для гликопротеинов тромбоцитов при ИТП аутоиммунного ответа. Более того, в работе R. Pellicano и соавт. показано отсутствие различий в продукции неспецифических аутоантител (антинуклеарных, антимикросомальных, антигладкомышечных) при ИТП у лиц с H. pylori и без них [16].
M. Kuwana и соавт. [17] предложили модель «патогенной петли» (a «pathogenic loop» model): появление антитромбоцитарных аутоантител у больных ИТП. Речь идет о том, что макрофаги ретикулоэндотелиальной системы «захватывают» тромбоциты через Fcγ-рецепторы и «передают» тромбоцитарные антигены (гликопротеиды) на Т-клетки (CD4+), которые в свою очередь, будучи активированными, стимулируют В-клетки на выработку антитромбоцитарных антител, связывающихся в результате с циркулирующими тромбоцитами, тем самым замыкая «патогенную петлю». После успешной эрадикации H. pylori антитромбоцитарные антитела удаляются [18], следовательно, «патогенная петля» остановлена. M. Kuwana и соавт., проведя проспективное исследование, показали повышенную фагоцитарную способность и низкие уровни экспрессии ингибирующего FcγRIIB в циркулирующих моноцитах у пациентов ИТП, инфицированных H. pylori, у H. pylori-негативных пациентов подобные изменения не выявлены. В случае успешной эрадикации наблюдалось подавление фенотипа активированного моноцита, затем уменьшался уровень антитромбоцитарных аутоантител и возрастало число тромбоцитов. Таким образом, H. pylori могут модулировать баланс Fcγ-рецепторов моноцитов/макрофагов в пользу активации рецепторов Fcγ [17, 19].
Целый ряд работ демонстрирует попытки определить особенности ИТП, ассоциированной с H. pylori, сравнивая клинические данные взрослых пациентов с ИТП с инфекцией H. pylori или без нее. Пациенты с ИТП, инфицированные H. pylori, были значительно старше, чем неинфицированные [20], но это может объясняться тем, что распространенность инфекции H. pylori с возрастом увеличивается среди населения в целом. Во многих других исследованиях не выявили существенных различий в любых других демографических или клинических характеристиках, включая пол, число тромбоцитов или ответ на терапию. В нескольких исследованиях обнаружены различия в генетических факторах пациентов с ИТП с H. pylori или без них. Итальянские исследователи D. Veneri и соавт. [21] изучали аллели HLA-DRB1 и DQB1 пациентов с ИТП и обнаружили, что у H. pylori-положительных пациентов были более низкие частоты DRB1*03 и более высокие частоты DRB1*11, DRB1*14 и DQB1*03 по сравнению с H. pylori-негативными пациентами. Однако у японских пациентов с ИТП связи между инфекцией H. pylori и аллелями HLA-DRB1 или DQB1 не выявлено, но обнаружено, что полиморфизм генов в локусах для интерлейкина-1β (ИЛ-1β) ассоциировался с инфекцией H. pylori у пациентов с ИТП в возрасте до 50 лет [17, 22]. Различий в уровнях ИЛ-2, -4 или -6 между пациентами с ИТП с инфекцией H. pylori и без нее не выявлено [23]. Сывороточные уровни хемокинов, как правило, регулируемые Т-клетками, были значительно выше у пациентов с ИТП с инфекцией H. pylori, чем у пациентов без нее, однако схожее повышение имело место также у лиц, у которых была H. рylori-ассоциированная патология желудочно-кишечного тракта, но не было ИТП [24]. Таким образом, к настоящему моменту не идентифицированы демографические, клинические, генетические или иммунологические характеристики, уникальные для пациентов с ИТП, инфицированных H. pylori. Вероятно, это связано с тем, что есть по меньшей мере две отдельные подгруппы пациентов с ИТП, инфицированных H. pylori: пациентов с вторичной ИТП, причиной которой служит H. pylori, в этом случае данные пациенты реагируют на эрадикационную терапию, а также с первичной ИТП и «случайной» сопутствующей инфекцией H. pylori.
Факторы, прогнозирующие положительное влияние эрадикационной терапии на течение ИТП, а именно на возрастание числа тромбоцитов, достаточно широко исследованы. Наиболее часто выявляемый благоприятный предиктор – более короткий анамнез ИТП [20, 25, 26], но в других исследованиях этой связи не находят [27–29]. Некоторые исследователи сообщают о значительном положительном эффекте эрадикации H. pylori у пациентов с легкой ТП, но у пациентов с тяжелой ТП был отмечен слабый ответ на эрадикацию [30]. Такие клинические характеристики, как возраст менее 65 лет при диагностике ИТП [25], более высокий исходный уровень тромбоцитов [25], отсутствие предшествовавшей терапии кортикостероидами [27], отсутствие сопутствующей терапии кортикостероидами [31, 32] и отсутствие предшествовавшей терапии для ИТП [25], также рассматриваются как факторы, прогнозирующие положительную «реакцию» тромбоцитов на лечение, однако сведения об их достоверности еще более противоречивы. В нескольких исследованиях изучалась генетическая предрасположенность к реакции тромбоцитов на антихеликобактерную терапию; в частности, показана ассоциация между гаплотипами HLA-DQB1*03 и более высокой вероятностью положительного ответа тромбоцитов [21]. Установлено, что одиночные нуклеотидные полиморфизмы в генах фактора некроза опухоли β и ингибитора Fcγ-рецептора IIВ (FcγRIIB) полезны для прогнозирования реакции ТП на эрадикационную терапию [32, 33]. Обнаружено, что наличие антитромбоцитарно специфичных анти-GPIb аутоантител предсказывает устойчивость к эрадикационной терапии H. pylori [17]. Противоречивы сведения о влиянии антител к CagA на прогноз повышения уровня тромбоцитов: исследование, проведенное в Италии, показало, что пациенты с ИТП и антителами к CagA чаще, чем пациенты без них, положительно реагируют на эрадикационную терапию [10], но исследование, проведенное в Японии, этого наблюдения не подтвердило [26]. R. Sato и соавт. [34] произвели оценку потенциальной связи ответа тромбоцитов на эрадикацию H. pylori с данными фиброэзофагогастродуоденоскопии и результатами гистологического исследования биоптатов желудка: выраженное воспаление и признаки атрофии в теле желудка расценены как предикторы благоприятного результата. Таким образом, и генетический фон, и бактериальные факторы, регулирующие уровень воспалительного ответа на инфекцию H. pylori, могут использоваться в целях прогнозирования успешности эрадикационной терапии для лечения ТП.
Несмотря на внушающие оптимизм данные исследований, ряд ученых сомневаются в наличии причинно-следственной связи между ИТП и инфекцией H. pylori, и, соответственно, влиянии эрадикационной терапии на число тромбоцитов. Так, результаты работы, проведенной A.D. Samson и соавт. [35], говорят об отсутствии существенных различий между H. pylori-позитивными и H. pylori-негативными пациентами в отношении числа тромбоцитов. Исследователи из Малайзии [36] сообщили о низкой распространенности инфекции H. pylori у пациентов с ИТП и отсутствии какого-либо значительного эффекта эрадикации H. pylori на число тромбоцитов.
ТП является распространенным гематологическим расстройством у пациентов с хроническим заболеванием печени [2, 37]. Распространенность ТП варьируется в зависимости от ряда факторов, таких как популяция пациентов и степень тяжести основного заболевания печени, а степень ТП служит ранним прогностическим маркером (индекс APRI – Aspartate-aminotransferase-to-Platelet Ratio Index), позволяющим делать предварительную оценку по вопросу наличия выраженного фиброза печени, не прибегая к биопсии [38]. Встречаемость ТП среди пациентов с циррозом печени достигает 80% [39].
Генез ТП при заболеваниях печени мультифакториален. Патофизиология ТП при хроническом заболевании печени уже давно [40] связана с гипотезой гиперспленизма, когда портальная гипертензия вызывает объединение и секвестрование всех корпускулярных элементов крови, преимущественно тромбоцитов, в расширенной и перегруженной селезенке. Пересадка печени нормализует ТП и уменьшает гиперспленизм [41].
Нарушение образования тромбоцитов при заболеваниях печени также связано со снижением активности и уровня тромбопоэтина – основного цитокина, продуцирующегося печенью и оказывающего воздействие на все стадии дифференцировки мегакариоцитов и синтеза тромбоцитов.
Помимо влияния на гемопоэтические клетки существует предположение о связывании тромбопоэтина непосредственно с циркулирующими в сосудистом русле тромбоцитами, что приводит к повышению их функциональной активности [42]. Снижение тромбопоэтинсинтетической функции печени приводит к уменьшению тромбопоэза в костном мозге и, следовательно, к ТП в периферической крови. Восстановление адекватной тромбопоэтинной продукции после трансплантации печени приводит к скорейшему восстановлению производства тромбоцитов [43].
Подавление образования тромбоцитов в костном мозге может быть вызвано алкоголем, одним из частых этиологических факторов заболевания печени. Алкоголь-индуцированная ТП обусловлена прямым токсическим эффектом алкоголя на мегакариоциты, что приводит к снижению производства, времени выживания и функции тромбоцитов [44, 45]. Ряд лекарственных средств, используемых в терапии заболеваний печени (азатиоприн, β-лактамные антибиотики и фторхинолоны, интерферон), потенциально могут вызывать лекарственно-индуцированную ТП, оказывая как прямое миелосупрессивное действие, так и иммуноопосредованное разрушение тромбоцитов. До недавнего времени схемы лечения пациентов с HCV (Hepatitis C Virus) включали интерферон, частым побочным эффектом которого является дозозависимая ТП, интерферон-индуцированная миелотоксичность и цитопения становились частой причиной прекращения терапии [2, 46].
Еще одним доказанным патогенетическим механизмом развития ТП являются аутоиммунные нарушения. Среди пациентов с хроническими заболеваниями печени различной этиологии до 64% имеют антитромбоцитарные антитела, которые в основном направлены против комплекса гликопротеина IIb-IX [47]. Наиболее часто иммуно-опосредованная ТП встречается при HCV, бактериальных и лекарственных поражениях печени. Аутоиммунные заболевания печени (аутоиммунный гепатит и первичный билиарный холангит – ПБХ) часто ассоциируются с другими аутоиммунными состояниями. Около 50% пациентов с ПБХ страдают по меньшей мере одним дополнительным аутоиммунным заболеванием, которое может включать ИТП [48].
Некоторые этиологические факторы поражения печени имеют собственные механизмы, влияющие на уровень тромбоцитов. Так, было показано, что HCV-инфекция может приводить к появлению ТП у пациента до появления стадии цирроза и гиперспленизма. Более 20 лет назад появились работы, свидетельствующие об ассоциации HCV и ТП [49–51]. До 30% пациентов с ИТП без признаков прогрессирующего заболевания печени серопозитивные по HCV [52]. Хроническая инфекция HCV может приводить к ТП с помощью различных механизмов [53], один из которых – прямое подавление костномозгового кроветворения [54]. Пациенты с HCV демонстрируют депрессию тромбоцитов даже в отсутствие спленомегалии [55] и нормализации уровня тромбоцитов после успешного лечения инфекции [56]. Известно, что хроническая HCV-инфекция связана с множеством аутоиммунных нарушений, около 40% пациентов с HCV-инфекцией имеют по меньшей мере одно иммуноопосредованное внепеченочное проявление в течение их болезни [57, 58]. Снижение уровня тромбоцитов при HCV коррелирует с тяжестью заболевания и сопровождается увеличением титров антитромбоцитарных Ig [49, 55]. Кроме того, HCV может напрямую взаимодействовать с тромбоцитами для связывания мембран тромбоцитов через множественные рецепторы клеточной поверхности, что в конечном итоге приводит к фагоцитозу тромбоцитов с антителом и ускоренному разрушению тромбоцитов ретикулоэндотелиальной системой [59].
Связывание HCV с тромбоцитами также может индуцировать образование антигенов на поверхности тромбоцитов или изменять свойства гликопродеидной мембраны тромбоцитов, что способствует формированию аутоантител, таких как GPIIb/IIIa, и последующему развитию ТП [60]. Наконец, HCV тесно связан с криоглобулинемией, а криоглобулины могут играть роль в формировании иммунных комплексов и развитии ТП [61].
Заключение
ТП представляет собой сложное и многофакторное явление, нередко встречающееся в практике врача-гастроэнтеролога. Глубокое понимание патофизиологии ТП в зависимости от причины ее возникновения имеет решающее значение при выборе стратегий лечения. Информированность специалистов о механизмах развития ТП при заболеваниях пищеварительной системы будет способствовать повышению успешности терапии.


