Введение
Муковисцидоз (МВ) – самое распространенное моногенное заболевание с аутосомно-рецессивным типом наследования [1]. Заболевание проявляется при наличии мутаций гена CFTR (Cystic fibrosis transmembrane conductance regulator – трансмембранный регулятор МВ). Аномальная работа гена приводит к дисфункции хлорного канала, расположенного в апикальной мембране экзокринных желез. В результате секрет желез становится чрезмерно вязким. При МВ чаще всего происходит поражение бронхолегочной системы с развитием бронхитов и формированием бронхоэктазов. Нарушение мукоцилиарного клиренса способствует присоединению патологической микрофлоры, что усугубляет течение болезни. Те же механизмы обусловливают формирование хронического синусита. Со стороны пищеварительной системы в поджелудочной железе (ПЖ) возможна блокада протоков, что приводит к синдрому мальабсорбции и задержке физического развития детей. В печени в результате сгущения желчи возможно формирование цирроза и желчнокаменной болезни. Кроме того, часто страдает репродуктивная система. Выраженность проявлений в различных органах разная, но, несмотря на это, тяжесть состояния, инвалидизация и смертность в подавляющем большинстве случаев обусловлены степенью поражения органов дыхания [1].
В настоящее время ген МВ достаточно хорошо изучен. Он располагается на длинном плече хромосомы 7, в области q31, имеет протяженность 250 тыс. пар нуклеотидных оснований, включает 27 экзонов, состоит из 2 нуклеотид-связывающих, 2 трансмембранных доменов и 1 центрального внутриклеточного регуляторного домена, функционирует как цАМФ-зависимый хлорный канал [2].
Выделено около 2000 различных мутаций и полиморфизмов гена CFTR. Мутации в гене CFTR разделяют на 5 классов в зависимости от нарушения функции белка. Мутации I–III классов («тяжелые») обусловливают глубокие нарушения функции проводимости хлорных каналов, IV–V классов («мягкие») протекают с частичным сохранением функции белка CFTR [3].
Генотип, включающий 2 мутации I–III классов, формирует «тяжелый» фенотип, ассоциированный с ранней панкреатической недостаточностью. При генотипе с наличием даже одной «мягкой» мутации формируется «мягкий» фенотип, для которого характерна сохранная функция ПЖ или позднее развитие панкреатической недостаточности [4].
В связи с внедрением скрининга новорожденных на МВ, совершенствованием генетических методов диагностики появилась возможность выявления большего количества мутаций в гене CFTR. При выявлении новых ранее неизвестных мутаций требуется изучение их фенотипических проявлений для определения их патологического влияния на течение заболевания [5].
Особенные затруднения возникают при описании фенотипических проявлений миссенс-мутаций и мутаций сайта сплайсинга. В отличие от «классических» мутаций со сдвигом рамки считывания или нонсенс-мутаций, при которых невозможность формирования нормального транскрипта определяет их очевидную «тяжесть», при миссенс-мутациях и мутациях сайта сплайсинга фенотипические проявления неочевидны. При выявлении таких мутаций бывают сложности с интерпретацией их влияния на течение болезни.
Постоянно ведутся поиски новых методов диагностики, подтверждающих дисфункцию CFTR [6].
Определенный интерес представляет мутация 3272-16Т>A, фенотипические проявления которой изучены недостаточно. Мутация 3272-16Т>A представляет собой сплайсинговую мутацию 19-го интрона гена трансмембранного регулятора МВ с заменой тимина на аденин в 16-м положении перед экзоном, начинающимся нуклеотидом с номером 3272. Впервые фенотипические проявления мутации были описаны у 10 взрослых пациентов в возрасте от 18,4 до 35 лет, имевших мутацию 3272-16 T>A в компаунде с другими мутациями. Были отмечены поздние сроки установления диагноза «муковисцидоз». У взрослых больных оставалась сохранной функция ПЖ, а также отмечались другие клинические проявления, подтверждающие «мягкий» фенотип [7, 8].
В настоящее время мутация 3272-16 T>A в гене CFTR внесена в стандартную панель 30 мутаций в лаборатории ДНК-диагностики ФГБНУ «Медико-генетический научный центр» РАМН. По данным Российского регистра 2014 г., мутация 3272-16 T>A встречалась в 0,34% аллелей (13-е место) в России: среди больных Москвы – 0,4% аллельной частоты (18-е место), Московской области – 0,35% (19-е место), Ярославской области – 1,22% (18-е место), в Челябинской области – 0,98% (12-е место), в Пермском крае – 0,83% (10-е место), Татарстан – 0,76% (10-е место), в Республике Удмуртия – 1,35% (6-е место), Мордовия – 8,33% (4-е место) [9].
Таким образом, мутация 3272-16 T>A встречалась у больных МВ в различных регионах России, преимущественно в Уральском и Приволжском федеральных округах [10]. В целом по стране в 2014 г. всего зарегистрировано 156 различных мутаций в гене CFTR, мутация 3272-16 T>A находилась на 17-м месте по частоте встречаемости. Фенотипические проявления данной мутации у детей не изучались.
Обобщенные данные по описываемой мутации могут дополнить компендиум человеческих генов и генетических фенотипов OMIM (Online Mendelian Inheritance in Man).
Целью нашей работы было изучение фенотипических особенностей у детей с МВ, носителей мутации 3272-16T>A.
Описание серии случаев
Всего в Пермском региональном центре муковисцидоза наблюдается 41 больной в возрасте до 18 лет. Мутация 3272-16 T>A была выявлена у 3 детей, больных МВ. Аллельная частота мутации 3272-16 T>A у детей, больных МВ, в Пермском крае составила 4,9%.
Приводим описание комплексного клинического обследования трех детей в возрасте 3, 8 и 9 лет с наличием в генотипе мутации 3272-16T>A.
У детей определяли антропометрические показатели роста и массы тела. Нутритивный статус оценивали с помощью индекса массы тела (ИМТ, кг/м2). Также рассчитывали массо-ростовой индекс (МРИ). За норму принимали показатели выше 90%.
Генетическое обследование проводили в лаборатории ДНК-диагностики ФГБНУ «Медико-генетический научный центр» РАМН (Москва). Проведен расширенный поиск частых мутаций в гене МВТР (CFTRdele 2,3 (del 21kb), F508del, 1507 del, 1677delA, 2143 del T, 2184 insA, 394 del TT, 3821 delT, L 138 ins, G 542 X, W1282X, N1303K, R334W, 3849+10kb C→T, 604 insA, 3944del TG, S1196X, 621+1 G→T, E92K, 3272-26A→G, 4015 delA, 4022 ins T, W1282R, 3272-16Т→А, 2785+5G→A, S466 X, 2785+5G→A, 3120+1G→A, R347P, S945L).
Исследование иммунореактивного трипсиногена (ИРТ) в пробах крови, высушенных в фильтровальной бумаге, проводили с помощью наборов DELFIA Neonatal IRT kit (Финляндия). За норму принимали пороговый уровень <99,5 центиля.
Для определения хлоридов в потовой жидкости применяли анализатор NANODUCT™ фирмы WescorInc. (США). Нормальными считались результаты менее 50 ммоль/л, 50–79 ммоль/л – сомнительными, более 80 ммоль/л – положительными.
Для исследования панкреатической эластазы-1 кала применяли тест-систему фирмы «Bioserv», Германия. Нормальными считались показатели выше 200 мкг/г кала.
Для оценки функции внешнего дыхания оценивали форсированную жизненную емкость легких (ФЖЕЛ) и объем форсированного выдоха за 1-ю секунду (ОФВ1).
У двух детей мутация 3272-16 T>A была выявлена в сочетании с тяжелыми мутациями 2143delT и F508del. Большой интерес представляет пациент, имевший мутацию 3272-16 T>A в гомозиготном состоянии.
У всех детей диагноз «муковисцидоз» был заподозрен в результате проведения скрининга новорожденных на МВ. Показатели ИРТ были высокими, при ретесте также показатели оставались выше нормы, что позволило заподозрить у детей МВ (табл. 1).

Потовый тест детям при постановке диагноза был проведен неоднократно, у всех пациентов отмечено значительное увеличение уровня хлоридов потовой жидкости. У одного больного, гомозиготного по мутации 3272-16 T>A, в возрасте 2 лет уровень хлоридов был 50 ммоль/л, что являлось верхней границей сомнительных результатов.
При сборе семейного анамнеза у всех детей семьи проживали на территории Пермского края. Населенные пункты, откуда родители пациента с гомозиготным носительством мутации 3272-16 T>A, находились в 7 км друг от друга, близкородственный брак родители отрицали.
У пациентки 9 лет с генотипом F508 del/3272-16T>A отмечено стойкое снижение нутритивного статуса, остальные больные в физическом развитии не отставали (табл. 2).

Показатели функции внешнего дыхания у больных в возрасте 8 и 9 лет были в пределах нормы (табл. 3). При проведении компьютерной томографии органов грудной клетки бронхоэктазов не выявлено.
У всех больных выявлены признаки синусита. У 8-летней пациентки отмечен полипозный синусит, по поводу которого проведено хирургическое лечение.
При обследовании пациентов на наличие экзокринной недостаточности ПЖ панкреатическая эластаза-1 кала была умеренно снижена у одного пациента. Однако у всех детей в копрограмме были отмечены незначительные признаки стеатореи. При проведении ультразвукового исследования (УЗИ) органов пищеварительной системы были выявлены изменения со стороны ПЖ, печени, аномалии со стороны желчного пузыря, признаки дисхолии (табл. 4).
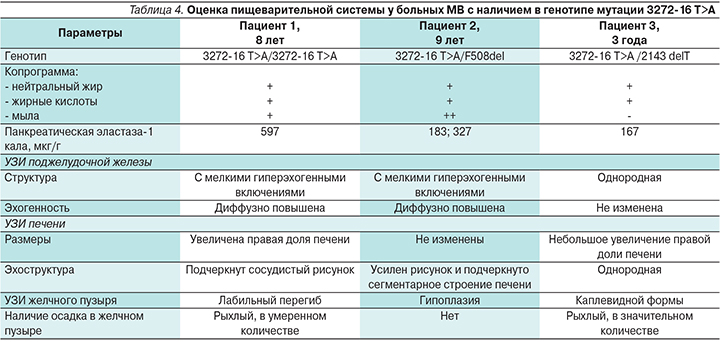
После установления диагноза было назначено комплексное лечение. Проведены муколитическая терапия, кинезотерапия, диетотерапия, заместительная ферментная терапия, витаминотерапия. Дети вели активный образ жизни, посещали детские дошкольные учреждения, школу. Одна пациентка занималась в детской танцевальной секции.
Обсуждение
У наблюдаемых нами детей, имевших в генотипе мутацию 3272-16 T>A, в школьном возрасте отмечалось поражение верхних дыхательных путей в виде хронических синуситов, в т.ч. с полипами. Со стороны бронхолегочной системы функция внешнего дыхания была сохранена, отсутствовали бронхоэктазы. Возможно, это было связано с проведением терапии с раннего возраста при установлении диагноза, а также с некоторым протективным влиянием «мягкого» генотипа.
Зафиксированы проявления относительной и слабовыраженной экзокринной недостаточности ПЖ у всех детей и снижение нутритивного статуса в одном случае. Однако дети при рождении имели высокие значения ИРТ, что указывало на влияние данной мутации на функцию ПЖ внутриутробно.
Как известно, «мягкие» мутации в ряде случаев характеризуются отрицательным потовым тестом, в частности такая ситуация нередка при cплайсинговой мутации 3849+10kbC>T [11]. В наших же случаях отмечено нарушение функции хлорных каналов в виде повышения хлоридов в потовой жидкости у всех троих пациентов. Течение заболевания у наблюдаемых больных в большей степени соответствовало описанию течения МВ при наличии в генотипе другой распространенной в России мутации – L138ins [12]. Однако сравнительную характеристику течения заболевания при этих мутациях проводить пока не представляется возможным в связи с дошкольным и младшим школьным возрастом наших пациентов. В то же время у описанных ранее 10 взрослых больных с наличием в генотипе мутации 3272-16T>A наблюдалась сохранная функция ПЖ и были все клинические проявления, подтверждающие «мягкий» фенотип МВ [8].
О функции хлорных каналов у взрослых больных при наличии мутации 3272-16T>A судить было сложно, т.к. потовый тест проводился не всем взрослым пациентам и диагноз «муковисцидоз» некоторым из них был установлен на основании клинических проявлений заболевания и генетического обследования.
Безусловно, вызывает интерес выявление у одного из наших пациентов гомозиготности по мутации 3272-16T>A. Гомозиготность по редкой мутации при исключении близкородственного брака говорит о возможно высоком популяционном носительстве таких мутаций в конкретном регионе. Что в свою очередь может подтверждать высказанный ранее тезис о высокой популяционной распространенности «мягких» мутаций гена CFTR в России по сравнению со странами Европы [10].
Отсутствие данных о выявлении мутации 3272-16T>A в странах Западной Европы и уже неоднократное обнаружение ее в России подтверждает, что мутация 3272-16T>A (наряду с мутациями L138ins, E92K) – патогномонична для России.
Заключение
Проведено комплексное клинико-лабораторное обследование трех детей с мутацией 3272-16T>A. Формирующийся фенотип с данной мутацией характеризовался классическим течением МВ, положительными показателями ИРТ и хлоридов пота, но при этом малой заинтересованностью в патологический процесс органов пищеварения, в частности ПЖ.
