
Введение
Синдром Дауна (СД) – хромосомный синдром, характеризующийся особым своеобразным фенотипом (близко расположенные глаза с монголоидным разрезом и эпикантом; большой, не помещающийся во рту язык; характерная форма носа), умственной отсталостью в сочетании с добродушным поведением, задержкой моторного развития, гипотонией, короткими конечностями (Бадалян Л.О., 1980; Кеннет Л. Джонс, 2011) [1, 2]. Частота встречаемости составляет, по данным разных авторов, от 1:800 до 1:1000 среди здоровых детей. По данным Л.О. Бадаляна и соавт. (1971), среди детей с психомоторными нарушениями частота встречаемости СД достигает 10% [1]. При СД отмечается экстракопирование генетического материала на 21-й хромосоме [1, 3].
Выделяют несколько вариантов, приводящих к развитию данного синдрома: трисомия 21-й хромосомы, транслокация, мозаицизм. Наиболее часто (более чем в 94 случаев) у пациентов с СД отмечается трисомия 21-й хромосомы с кариотипом 47-й. Транслокационный синдром (45,XX, t(14;21q)) занимает второе место по частоте и встречается в 1,2–4,0% случаев. Мозаицизм (46,XX/47,XX,+21) выявляется в 1,0–2,4% случаев [2, 4].
Эпилепсия у пациентов с СД, по данным разных авторов, может выявляться в 8–9% случаев [2, 5, 6]. Среди больных СД, страдающих эпилепсией, синдром Веста (СВ) встречается в 49–56% случаев [4, 7]. При этом, по данным Kurokawa и соавт. (1980), среди больных СВ пациенты с СД составляют всего 1% случаев [8]. Вместе с тем Stafstrom, Konkol (1994) отмечают, что эпилептические спазмы (ЭС) при СД встречаются в 8–10 раз чаще, чем в общей популяции [9].
СВ – возраст-зависимый полиэтиологический эпилептический синдром, относящийся к группе младенческих эпилептических энцефалопатий.
СВ характеризуется следующими критериями [10, 11]:
- Особый тип эпилептических приступов – эпилептические (инфантильные) спазмы, представляющие массивные миоклонические и (или) тонические, про- и (или) ретропульсивные, симметричные и (или) асимметричные, серийные и (или) изолированные спазмы аксиальной и конечностной мускулатуры.
- Изменения на электроэнцефалограмме в виде гипсаритмии.
- Задержка психомоторного развития.
По этиологии, особенностям течения и прогнозу у пациентов с трисомией по 21-й хромосоме выделяют идиопатический и криптогенный/симптоматический варианты СВ. Согласно Silva и соавт. (1996), основные диагностические критерии идиопатического варианта СВ у пациентов с СД: отсутствие врожденных кардиопатий и признаков последствия перинатальной гипоксически ишемической энцефалопатии [12]. Для идиопатического варианта СВ при СД характерны симметричные спазмы. В единичных случаях спазмы могут быть асимметричными. При идиопатическом варианте СВ на электроэнцефалограмме (ЭЭГ) регистрируется классическая симметричная гипсаритмия [13].
По данным Stafstrom, Konkol (1994), наиболее частой причиной возникновения симптоматического варианта СВ у пациентов с трисомией 21-й хромосомы является последствие перинатальной гипоксически-ишемической энцефалопатии [9]. Escofet и соавт. (1995), Smigielska-Kuzia и соавт. (2009) отмечают важность выявления при симптоматическом варианте СВ латерализационных признаков, которые могут указывать на структурное локальное повреждение головного мозга. К этим признакам можно отнести асимметричные тонические спазмы в виде формирования «позы фехтовальщика», отведения головы и глаз в сторону. Нередко при симптоматическом варианте СВ отмечаются фокальные приступы, версивные или тонические. На ЭЭГ при симптоматическом варианте, как правило, регистрируется «модифицированная» гипсаритмия с преобладанием региональных или мультирегиональных эпилептиформных нарушений [14, 15].
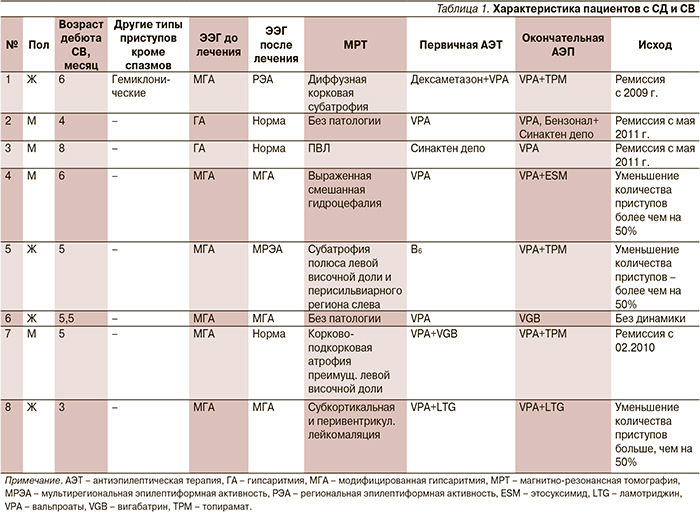
Вне зависимости от этиологии после дебюта ЭС у большинства пациентов с СД наблюдается выраженная задержка или регресс психомоторного развития. При видео-ЭЭГ-мониторинге ЭС при СД характеризуются классическим ЭЭГ-паттерном: появлением высокоамплитудной билатерально-синхронной диффузной дельта-волны, которая «прерывает» картину гипсаритмии, с присоединением низкоамплитудной быстроволновой активности (lafa), сменяемой или сопровождаемой коротким эпизодом уплощения биоэлектрической активности; далее постепенно наблюдается восстановление картины гипсаритмии [12].
В отечественной литературе сочетание СД и СВ описывается в единичных публикациях [16–18]. С целью изучения анамнестических, клинических, электроэнцефалографических и нейровизуализационных особенностей у пациентов СВ и СД было проведено настоящее исследование.
Материал и методы
В исследование были включены 8 пациентов с генетически верифицированным СД и СВ, которые проходили обследование и лечение на клинической базе Института детской неврологии и эпилепсии им. Святителя Луки с 2006 по 2015 г.
Диагностика СВ проведена согласно критериям международной классификации эпилепсий, эпилептических синдромов и схожих заболеваний (1989), а также на основании доклада комиссии ILAE по классификации и терминологии (2001).
Всем пациентам было проведено неврологическое обследование; рутинное ЭЭГ-исследование; также во всех случаях проводился продолженный видео-ЭЭГ-мониторинг (ВЭМ) с включением сна (аппарат электроэнцефалограф-анализатор ЭЭГА-21/26 «ЭНЦЕФАЛАН-131-03», модификация 11, Медиком МТД; видео-ЭЭГ-мониторинг «Нейроскоп 6.1.508», Биола); была проведена магнитно-резонансная томография (магнитно-резонансная система Sigma Infinity GE с напряжением магнитного поля 1,5 Тесла).
В большинстве случаев СД был заподозрен при проведении пренатального скрининга, инвазивные исследования не проводились. Диагноз был подтвержден в постнатальном периоде методом кариотипирования в 7 случаях и при помощи хромосомного микроматричного анализа в 1 случае. У всех пациентов наблюдалась трисомия по 21-й хромосоме.
Исследование имело ретроспективный характер. Оценивались возраст дебюта приступов, их семиология и частота, проводился анализ клинико-электроэнцефалографических коррелятов, лечения антиэпилептическими препаратами (АЭП), а также клинических и электроэнцефалографических исходов заболевания (табл. 1).
Результаты
Возраст дебюта приступов варьировался в диапазоне от 4 до 8 месяцев, в среднем составив 5,6 месяца. Длительность наблюдения – до 7 лет. У всех пациентов в разгар заболевания приступы протекали серийно, частота варьировалась от 5 до 120 приступов за сутки.
У всех пациентов, по данным анамнеза, еще до появления ЭС наблюдалась характерная для СД задержка психомоторного развития. Однако после дебюта ЭС имел место регресс в виде утраты ранее приобретенных навыков, а после купирования приступов или уменьшения их частоты – прогресс, в одном случае – даже значительный скачок в развитии. Клинические проявления ЭС варьировались от едва заметных кивков и клевков, захватывавших мышцы шеи, до массивных сокращений, вовлекавших в приступ большие группы мышц.
При видео-ЭЭГ-мониторинге у шести пациентов зарегистрированы как одиночные (в трех случаях), так и серийные (в пяти случаях) ЭС. Наблюдались флексорные (в четырех случаях), экстензорные (в двух случаях), а также флексорно-экстензорные (в двух случаях) приступы. В двух случаях выявлены латерализационные признаки в виде отведения головы и глаз в сторону.
В большинстве случаев инфантильные спазмы наблюдались как единственный тип пароксизмов. И только у одного пациента помимо ЭС регистрировались гемиклонические приступы. Характерно, что у данного пациента на МРТ выявлена корково-подкорковая атрофия с асимметричным поражением – преимущественно в области левой височной доли.
Изменения на ЭЭГ
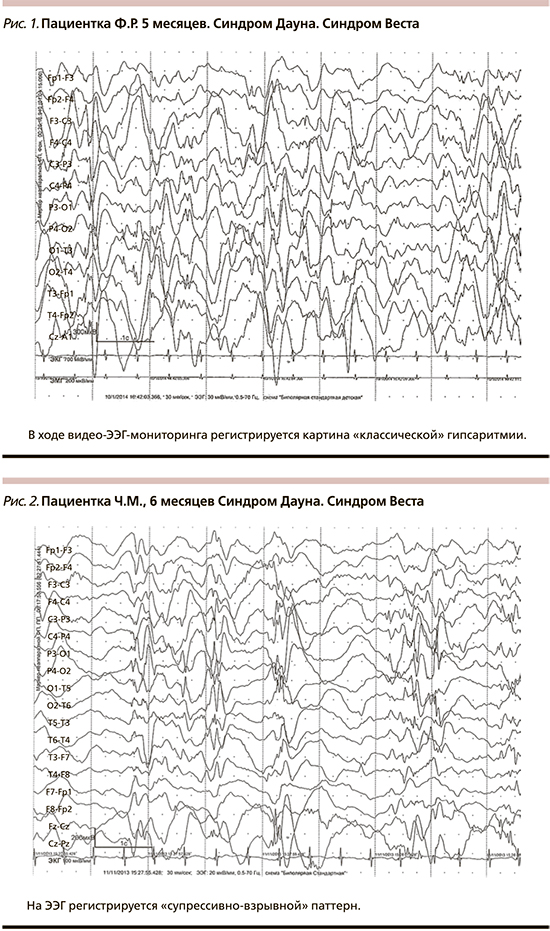
В ходе продолженного видео-ЭЭГ-мониторинга во всех случаях были выявлены эпилептиформные изменения на ЭЭГ. У всех пациентов в разгар заболевания была отмечена гипсаритмия (рис. 1). При этом в двух случаях наблюдался классический вариант, в шести – констатировалась модифицированная гипсаритмия: в двух случаях с персистированием «супрессивно-взрывного» паттерна (рис. 2), в трех из них – зонально акцентуированная с затылочной (рис. 3) и лобной акцентуацией. У одного пациента на фоне гипсаритмии выявлена устойчивая региональная эпилептиформная активность в височной области.

В ходе исследования были выявлены следующие ЭЭГ-паттерны ЭС:
- диффузная дельта-волна, которая «прерывает» картину гипсаритмии, сменяемой коротким эпизодом уплощения биоэлектрической активности;
- диффузная дельта-волна в сочетании с низкоамплитудной быстроволновой активностью (lafa), сменяемая коротким эпизодом уплощения биоэлектрической активности.
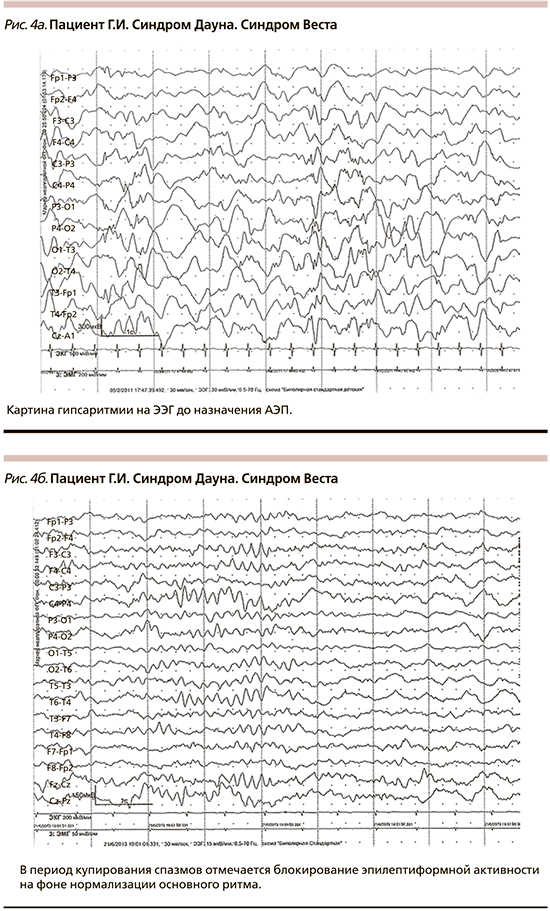
Данные ЭЭГ могли претерпевать изменения на фоне АЭТ. В шести случаях в период АЭТ наблюдалось блокирование картины гипсаритмии на ЭЭГ (рис. 4а, 4б). Из них в трех случаях после купирования приступов на ЭЭГ регистрировался вариант возрастной нормы, эпилептическая активность не была зарегистрирована. Кроме того, у троих пациентов на фоне лечения биоэлектрическая активность соответствовала возрастной норме, при этом у одного из них регистрировалась региональная и у двоих мультирегиональная эпилептиформная активность.
В остальных случаях (2 пациента) гипсаритмия сохранялась.
Нейровизуализация
При проведении нейровизуализации в 2 случаях изменения не выявлены, в остальных выявлены диффузная корковая субатрофия (1 пациент), диффузная корково-подкорковая атрофия (1 случай), корково-подкорковая атрофия с преимущественным вовлечением левой височной доли (1 случай), перивентрикулярная лейкомаляция (2 случая), субатрофия полюса левой височной доли и перисильвиарного региона (1 случай).
Терапия
При назначении АЭТ улучшение наблюдалось в 87,5% случаев (ремиссия или уменьшение количества приступов на 50% и более). При этом устойчивая ремиссия была достигнута в 4 случаях (50%), причем в 3 из них – клинико-электроэнцефалографическая. В группе пациентов с достигнутой ремиссией в одном случае применялась монотерапия, в остальных – политерапия. Еще в трех случаях отмечено уменьшение частоты приступов более чем на 50% (37,5% случаев). Лишь только у одной пациентки при назначении АЭТ не отмечено положительной динамики.
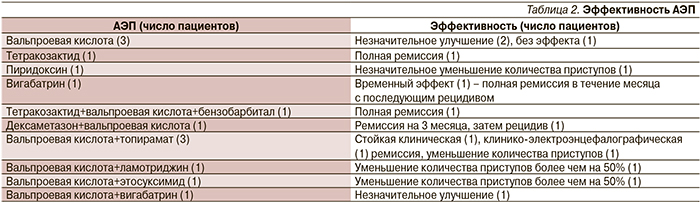
В лечении СВ пациенты нашей группы применяли различные АЭП и их сочетания (табл. 2).
Вальпроевая кислота применялась в качестве монотерапии в трех случаях, в двух из которых наблюдалось небольшое улучшение и в одном – без эффекта. Препараты вальпроевой кислоты применялись также в политерапии (в 8 случаях): одном – в сочетании с тетракозактидом и бензобарбиталом; в результате достигнута полная ремиссия. В трех случаях производные вальпроевой кислоты назначались в комбинации с топираматом: у двух пациентов достигнута стойкая клинико-электроэнцефалографическая ремиссия, в одном – отмечено уменьшение количества приступов более чем на 50%. При сочетании вальпроатов с ламотриджином или этосуксимидом наблюдалось уменьшение количества приступов более чем на 50% (2 пациента). Совместное применение вальпроатов с вигабатрином привело лишь к небольшому улучшению (1 пациент).
В сочетании с курсовым приемом дексаметазона была достигнута ремиссия на 3 месяца, затем – рецидив приступов.
Тетракозактид (синтетический аналог адренокортикотропного гормона) назначался в двух случаях (в одном – в качестве монотерапии и в политерапии – в другом (в сочетании с вальпроевой кислотой и бензобарбиталом). Во всех двух случаях отмечено достижение полной клинико-электроэнцефалографической ремиссии.
Пиридоксин назначен в одном слу-чае – в качестве монотерапии с временным положительным эффектом в виде уменьшения количества приступов.
Вигабатрин в качестве монотерапии назначен в одном случае с незначительным временным эффектом. В сочетании с вальпроевой кислотой также отмечен небольшой положительный эффект в виде уменьшения количества приступов (1 пациент).
Топирамат назначен в комбинации с вальпроевой кислотой как добавочный препарат (3 пациента): в 2 случаях достигнута стойкая клинико-электроэнцефалографическая ремиссия и еще в 1 – уменьшение количества приступов более чем на 50%.
Ламотриджин также назначался в сочетании с вальпроевой кислотой одному пациенту, уменьшение количества приступов наблюдалось более чем на 50%.
Этосуксимид в сочетании с вальпроевой кислотой применен одним пациентом: наблюдалось уменьшение количества приступов более чем на 50%.
Cочетание дексаметазона с вальпроевой кислотой привело к ремиссии на 3 месяца, после которой произошел рецидив эпилептических приступов.
Таким образом, наше исследование показало, что наибольший эффект наблюдался при назначении тетракозактида как в моно-, так и в политерапии, а также вальпроевой кислоты в комбинации с топираматом.
Обсуждение
ЭС при СД дебютируют, по данным разных авторов, в диапазоне от 3 до 18 месяцев, в среднем в 7–8 месяцев [12, 13]. Наши данные соответствуют таковым литературы. У представленных нами пациентов возраст дебюта ЭС варьировался в диапазоне от 4 до 8 месяцев, в среднем составив 5,6 месяца.
Анализируя полученные результаты клинико-электроэнцефалографи-ческих особенностей пациентов нашей группы, можно отметить, что характер приступов (ЭС), картина на ЭЭГ (гипсаритмия или модифицированная гипсаритмия), ЭЭГ-паттерн пароксизмов в целом не отличались от симптоматики СВ другой этиологии. Основной триадой симптомокомплекса СВ как в нашем исследовании, так и по данным других авторов служили ЭС, гипсаритмия на ЭЭГ и задержка психомоторного развития [12, 20, 21]. Представляет интерес, что в нашем исследовании в 6 случаях констатировалась именно модифицированная гипсаритмия.
Традиционно СВ у пациентов с хромосомной патологией относят к симптоматической эпилепсии, которая, по мнению большинства эпилептологов, является прогностически неблагоприятной в отношении приступов и когнитивных функций. Действительно, при проведении нейровизуализации лишь у двух пациентов не были выявлены изменения. В остальных случаях на МРТ констатировались признаки перинатальной гипоксически ишемической энцефалопатии. Однако высокая эффективность АЭТ в отношении приступов (несмотря на изменения на МРТ) у пациентов нашей группы заставляет согласиться с мнением некоторых авторов, согласно которому СВ у пациентов с СД может протекать как идиопатический вариант. Так, французская группа ученых в 1996 г. сформулировала основные диагностические критерии идиопатического варианта СВ у пациентов с СД: отсутствие врожденных кардиопатий и признаков последствий перинатальной гипоксически ишемической энцефалопатии [12]. С учетом отсутствия последствий перинатальной гипоксически ишемической энцефалопатии при нейровизуализации, врожденных кардиопатий у некоторых из наших пациентов, зафиксированных у них симметричных инфантильных спазм, а также классического варианта гипсаритмии на ЭЭГ эти случаи можно было бы отнести к идиопатическим. Однако это верно только в отношении СВ у больных СД. В остальных случаях СВ термин «идиопатический вариант» носит иное смысловое значение, крайне редок и признается не всеми авторами [27].
Назначение АЭП позволило получить значительное улучшение (в 87,5% случаев) СВ при СД, при этом была достигнута ремиссия у 50% пациентов. Это согласуется с данными Stafstrom, Konkol (1994), показавших, что ЭС у пациентов с СД нередко лучше поддаются лечению по сравнению с симптоматическим СВ в общей популяции [9]. Наше исследование показало, что эффективность АЭП в отношении купирования ЭС у пациентов с СД сопоставима с результатами лечения СВ в популяции в целом. Медикаментозная ремиссия приступов при СВ (без учета этиологии) составляет от 33 до 80%, по данным различных авторов [22, 21]. Согласно нашему предыдущему исследованию, посвященному терапии СВ, ремиссия достигается в 50% случаев [19].
Высокая эффективность в лечении инфантильных спазмов у детей с СД при применении адренокортикотропного гормона (АКТГ) и его синтетических аналогов показана различными авторами [4, 14, 23, 24]. В нашем исследовании синтетический аналог АКТГ применен в двух случаях, в результате была достигнута стойкая клинико-электроэнцефалографическая ремиссия. У остальных пациентов ремиссии (в двух случаях) и значительного уменьшения количества приступов (в трех случаях) удалось достичь на фоне лечения вальпроевой кислотой в сочетании с топираматом, ламотриджином, а также этосуксимидом. В одном из этих случаев первоначально ремиссия наступила на фоне терапии дексаметазоном в сочетании с вальпроевой кислотой, однако через 3 месяца произошел рецидив.
По данным Nabbout и соавт. (2001), высокоэффективен вигабатрин. При применении вигабатрина коротким курсом в четырех случаях из пяти больных СВ и СД отмечали полное купирование инфантильных спазмов, причем у трех из них уже в течение первой недели лечения. Курс применения вигабатрина составил 6 месяцев после наступления ремиссии, после чего его отменили. Длительный катамнез показал отсутствие рецидивов приступов. Авторы считают оправданным применение вигабатрина коротким курсом в связи с отличным эффектом, а также с целью снижения вероятности развития побочных эффектов со стороны зрения [13]. Caraballo и соавт. (1998) при назначении вигабатрина 15 больным СВ и СД констатировали купирование инфантильных спазмов в 10 случаях в течение первых двух недель после начала терапии и еще у 4 пациентов в течение 6 месяцев [23]. Высокая эффективность вигабатрина отмечается и в других публикациях, посвященных лечению инфантильных спазмов при СД [24–26]. В нашем исследовании вигабатрин в качестве монотерапии применен в одном случае, ремиссии или уменьшения количества приступов достичь не удалось. Однако оценка данного случая не представляется возможной, поскольку пациенты прекратили наблюдение у лечащего врача, не закончив курса, по неизвестным причинам. Еще в одном случае вигабатрин применялся в сочетании с вальпроевой кислотой с небольшим положительным эффектом.
Интересной представляется публикация аргентинской группы ученых Caraballo и соавт. (2004), показавших хороший эффект применения высоких доз пиридоксина в лечении инфантильных спазмов у больных СД [23]. В исследование были включены 7 пациентов с СВ и СД, которым в схему терапии был добавлен пиридоксин. Назначался пиридоксин перорально в дозе 200–400 мг/сут (25–50 мг/кг/сут). Купирование приступов и нормализация ЭЭГ были отмечены в 5 из 7 случаев, причем длительный катамнез показал отсутствие рецидивов в этой группе. Кроме того, положительным моментом стало отсутствие побочных эффектов при применении пиридоксина. В другом исследовании показан примерно одинаковый эффект при назначении пиридоксина, АКТГ и вигабатрина, однако применение последних двух препаратов вызвало значительно больший процент побочных эффектов [23]. Однако данное исследование единичное и его недостаточно, чтобы делать серьезные выводы об эффективности пиридоксина.
Наше исследование показало высокую эффективность комбинации вальпроатов с топираматом. Из трех случаев назначения данной схемы двум пациентам достигнута стойкая клинико-электроэнцефалографическая ремиссия, в одном случае отмечено уменьшение количества приступов более чем на 50%. Мы не нашли публикаций, посвященных изучению эффективности вальпроатов в комбинации с топираматом в терапии СВ у пациентов с СД. Полученные результаты, по нашему мнению, крайне важны для российских эпилептологов, т.к. производные вальпроевой кислоты и топирамат зарегистрированы на территории Российской Федерации в отличие от вигабатрина.
С учетом особенностей российского медицинского законодательства и труднодоступности препаратов с доказанной эффективностью (таких как АКТГ и вигабатрин), а также с учетом результатов настоящего исследования мы предлагаем следующий порядок назначения АЭП при лечении СВ у пациентов с СД:
- Пиридоксин;
- При недостаточной эффективности – вальпроевая кислота в сиропе или микрогранулах;
- При недостаточной эффективности к терапии добавить топирамат в микрогранулах;
- В отсутствие достижения ремиссии – курсом тетракозактид;
- В качестве резервного препарата может рассматриваться вигабатрин.
Клиническая оценка действия каждого препарата должна быть очень быстрой (около недели), т.к. каждый день персистирования спазмов наносит непоправимый вред когнитивным функциям пациента [7].
Несмотря на хороший контроль приступов при адекватно подобранной терапии СВ, психомоторное развитие больных СД происходит с выраженным отставанием в подавляющем большинстве случаев. Как указывают Goldberg-Stern, 2001 [5], O. Sanmaneechai,2013 [27], задержка связана с основным заболеванием. Это подтверждают и данные нашего исследования: хотя после купирования приступов наблюдался прогресс в психическом и двигательном развитии, все же оно происходило с выраженной задержкой.


