
Введение
Врожденный гипопитуитаризм – редкое заболевание с распространенностью от 1/4000 до 1/10 000 [1]. Врожденный гипопитуитаризм может характеризоваться дефицитом функции одного гормона, чаще всего изолированным дефицитом гормона роста (ИДГР) или дефицитом двух и более гипофизарных гормонов (множественный дефицит гормонов гипофиза – МДГГ). В 5–16% случаев врожденного гипопитуитаризма идентифицируется генетическая основа заболевания [2]. Как правило, мутации возникают в генах, ответственных за онтогенез гипоталамо-гипофизарной области. Однако факт того, что этиология заболевания чаще всего остается неизвестной, позволяет предположить важную роль и других генов в развитии заболевания [3].
Гипофиз – это центральный регулятор, ответственный за контроль роста, метаболизм, развитие, репродукцию и поддержание гомеостаза путем регуляции периферических эндокринных желез в организме. Несмотря на свою главенствующую роль в эндокринной системе организма, гипофиз имеет довольно простую организацию. Ранее ошибочно полагали, что гипофиз представляет собой смесь клеток, но в процессе детального изучения обнаружили, что клетки гипофиза собраны в трехмерные структуры и каждая такая структура выполняет четко определенную роль. Передняя (и выделяемая отдельно в зарубежных источниках как промежуточная) доля гипофиза происходит из оральной эктодермы, в то время как задняя доля – из отростка воронки промежуточного мозга. Развитие гипофиза, а также детерминация и спецификация клеток зависят от экспрессии и взаимодействия сигнальных молекул и транскрипционных факторов в различных, но перекликающихся пространственных и временных паттернах (см. рисунок) [1].
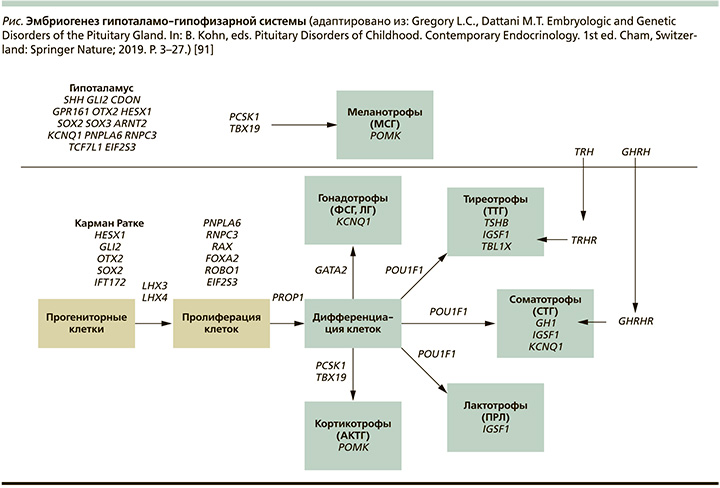
В результате этого взаимодействия формируется пять высокодифференцированных типов клеток, среди которых соматотрофы (вырабатывают СТГ – соматотропный гормон), кортикотрофы (синтезируют АКТГ – адренокортикотропный гормон), тиреотрофы (вырабатывают ТТГ – тиреотропный гормон), гонадотрофы (синтезируют ЛГ – лютеинизирующий и ФСГ – фоликуллостимулирующий гормоны), а также лактотрофы (вырабатывают ПРЛ – пролактин).
С момента картирования первого гена, участвовавшего в развитии гипофиза у человека в 1992 г., существенно расширилось и изменилось понимание генетических основ гипопитуитаризма: за последние 3 десятилетия обнаружены патогенные варианты более 30 генов [4, 5]. Применение современных методов молекулярно-генетического тестирования позволяет идентифицировать все новые гены, участвующие в развитии гипофиза, а также расширять знания о фенотипах, связанных с ранее известными генами.
В таблице представлены гены, мутации в которых ассоциированы с развитием врожденного гипопитуитаризма.
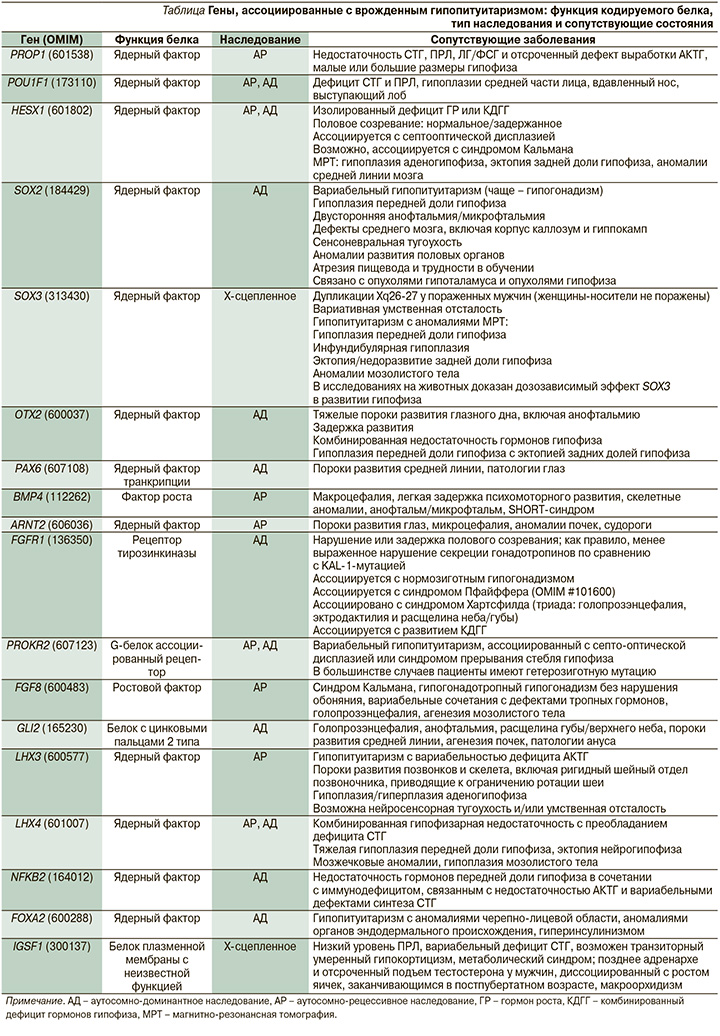
Мутации в генах, участвующих в ранних этапах развития гипоталамо-гипофизарной области (HESX1, LHX3, LHX4, SOX2, SOX3, GLI2, OTX2), связаны со структурными аномалиями не только гипоталамо-гипофизарной оси, но и срединной линии, c внегипофизарными дефектами. Мутации же в генах, экспрессирующихся на более поздних этапах развития (PROP1 и POU1F1), связаны с фенотипом заболевания без экстрагипофизарных дефектов [6].
В неонатальном периоде клинические проявления врожденного гипопитуитаризма неспецифичны и включают апноэ, летаргию, плохой набор массы тела, плохой аппетит; определяются этиологией, возрастом манифестации, а также количеством и типом затронутых тропных гормонов [1, 5, 7].
Задержка роста при дефиците СТГ манифестирует чаще после первого года рождения, реже может проявляться уже в младенчестве. Гиперинсулинемическая тяжелая гипогликемия, связанная с судорожным синдромом, сохраняющаяся более 3 дней и ведущая потенциально к развитию повреждения головного мозга, – наиболее частое проявление дефицита СТГ, а также АКТГ [8]. При этом сочетание дефицита этих тропных гормонов приводит к более тяжелым и частым эпизодам гипогликемии. Дефицит АКТГ характеризуется также гипонатремическими и гипотензивными проявлениями. Признаки дефицита ЛГ в младенчестве – это микропения (SD полового члена менее -2,5 от среднего значения) и крипторхизм, т.к. именно ЛГ отвечает за выработку тестостерона плодом, а также за процесс опускания яичек. Микропения выявляется также при ИДГР, что говорит о большом значении СТГ для роста полового члена во внутриутробном и постнатальном периодах [9, 10].
Неспецифичным, но довольно частым проявлением гипопитуитаризма в неонатальном периоде являются холестатическая желтуха и неонатальный гепатит (что ряд авторов объясняют дефицитом глюкокортикоидов и тиреоидных гормонов) [11]. При этом уровни трансаминаз и γ-глутамилтранспептидазы у пациентов в пределах референса.
Сложность диагностики заболевания состоит в том, что мутации в разных генах могут приводить к формированию схожих фенотипов и при этом одна и та же мутация может быть связана с разными фенотипами даже у членов одной семьи. Генетическое исследование имеет основополагающее значение для понимания тонкостей органогенеза гипофиза, возможности ранней, в идеале – доклинической, диагностики заболевания, а также прогнозирования прогрессирования заболевания и последующего наблюдения за пациентами.
Редкие причины септооптической дисплазии (СОД, синдром Де Морсье)
Это редкое заболевание с распространенностью около 1/10 000 живорожденных с соотношением мужчин и женщин 1:1 [12]. Впервые оно было описано у 7-месячного ребенка с отсутствием прозрачной перегородки и аномалиями зрительного нерва [13]. В дальнейшем стало известно, что СОД характеризуется наличием двух из следующих признаков: аплазия или гипоплазия прозрачной перегородки, аплазия или гипоплазия зрительных нервов, перекреста зрительных нервов, гипоплазия гипофиза с развитием гипопитуитаризма [14].
СОД – это этиологически гетерогенная группа заболеваний, наиболее частой причиной является мутация в гене HESX1, схожий фенотип наблюдается также при мутациях в генах SOX2, SOX3, OTX2 [15]. Эти гены экспрессируются в регионах, которые определяют формирование переднего мозга и связанных с ним срединных структур, таких как гипоталамус и гипофиз, и поэтому мутации в этих генах связаны с заметной фенотипической гетерогенностью [16–21].
В 1996 г. E. Hermes et al. выделили новый ген гомеобокса кармана Ратке, а в 1998 г. M.T. Dattani et al. картировали ген HESX1 (OMIM 601802) и определили, что он содержит 4 экзона [22]. Они описали гомозиготную мутацию R160C у родных брата и сестры от кровнородственного брака с клиническими проявлениями СОД – аплазией мозолистого тела, межжелудочковой перегородки и эктопией задней доли гипофиза [16]. Впоследствии был описан ряд аутосомно-рецессивных и доминантных мутаций с широким разнообразием фенотипов.
OTX2 (OMIM 600037)
OTX2 – это ген семейства гомеобокса, расположенный на 14-й хромосоме и состоящий из 5 экзонов, первые два из которых некодирующие [23]. Мутации в данном гене наследуются аутосомно-доминантно и характеризуются неполной пенетрантностью.
В первую очередь OTX2 участвует в развитии глаз, а также мозга и лицевой части черепа. Известно по меньшей мере 10 патологических гетерозиготных вариантов в гене OTX2, идентифицированных у пациентов с пороками развития глаз [17, 24]. Также OTX2 экспрессируется в гипофизе и выполняет функцию трансактиватора промоторов POU1F1 и HESX1, а также промотора IRBP (интерстициального ретиноид-связывающего белка), участвующего в функции глаза [25, 26]. Экспрессируясь в гипоталамусе, OTX2 выполняет функцию трансактиватора GNRH1, необходимого для секреции гонадотропин-рилизинг-гормона гипоталамусом. То есть GNRH1 также является геном-мишенью OTX2 [27].
Глазной фенотип варьируется от анофтальмии/микрофтальмии до нормального развития глаз даже у членов одной семьи, характерно поражение глазного нерва – гипоплазия/дисплазия/аплазия. С учетом доказанной роли гена в развитии гипофиза помимо чистого глазного фенотипа описывают сочетание глазного фенотипа с гипопитуитаризмом и очень редко – изолированный гипопитуитаризм [28]. Клиника гипопитуитаризма чаще характеризуется изолированным дефицитом СТГ, но не ограничивается им, есть описания пациентов с пангипопитуитаризмом, с дефицитом ТТГ, АКТГ и СТГ и характерной гипоплазией гипофиза по данным МРТ головного мозга.
Характерными, но необязательными для синдрома также являются задержка умственного развития (от легкой до тяжелой), эпилептические приступы, нейросенсорная тугоухость, пороки костной системы (клинодактилия, дисгнатия и др.) [24, 29, 30].
GLI2 (OMIM 165230)
GLI2 – большой полиморфный ген, расположенный в положении q14 на длинном плече хромосомы 2, содержащий 13 экзонов и кодирующий белок, содержащий 1586 аминокислот. Белок GLI2 – фактор транскрипции, содержащий ДНК-связывающий домен с цинковым пальцем. Характер наследования заболевания аутосомнодоминантный, заболевание носит название «синдром Каллера–Джонса». Клинический фенотип крайне гетерогенен и в первую очередь характеризуется гипопитуитаризмом (от ИДГР до КДГГ) с постаксиальной полидактилией или без нее, лицевым дисморфизмом и умственной отсталостью [31]. Y. Zhang et al. сообщили о необычном случае синдрома Каллера–Джонса вследствие ранее не описанной мутации у мальчика 15 лет, в течение 10 лет наблюдавшегося с аносмией с подозрением на синдром Кальмана, а также с болью в придатках яичка с 14 лет. Молекулярно-генетическое исследование позволило верифицировать диагноз [32]. Имеются данные о нескольких гетерозиготных вариантах GLI2, которые встречаются также в базе данных здоровых людей. Остается неясным, все ли варианты имеют клиническое значение. Неполная пенетрантность также указывает на то, что одной мутации, вероятно, недостаточно, чтобы развить фенотип, и она должна быть связана с другими генетическими факторами и/или факторами окружающей среды.
LHX3 (OMIM 600577)
LHX3 – ген, кодирующий фактор транскрипции домена LIM, участвующего в ранних этапах онтогенеза гипофиза. Мутации в этом гене приводят к развитию аутосомно-рециссивного заболевания, характеризующегося гипопитуитаризмом, при этом патогномонично развитие дефицита СТГ и ТТГ, у двух третей развивается дефицит ЛГ/ФСГ и ПРЛ, в то время как только у трети встречается дефицит АКТГ [33]. Доказано, что гетерозиготные по LHX3 пациенты развивают более мягкий фенотип гипопитуитаризма, чем гомозиготные. Это крайне важно, т.к. до недавнего времени считалось, что пациенты с гетерозиготными мутациями не подвержены риску развития гипофизарной недостаточности. Менее половины пациентов имеют гипоплазию аденогипофиза при нормальных размерах и расположении нейрогипофиза.
Внегипофизарные проявления довольно распространены, включают ригидность шеи, увеличенные в размерах роднички, нарушение слуха, вентральное смещение или реже – истончение мозолистого тела и долихоцефалию [34–37].
Было опубликовано описание семьи, в которой в одном поколении наблюдались два выкидыша, рождение одного ребенка со сложными гетерозиготными вариантами LHX3 p.C118Y и c.252-3 C>G с КДГГ и ограниченной ротацией шеи. Члены той же семьи, несущие нарушение сплайсинга c.252-3 C>G, имели высокую частоту встречаемости ригидности шеи [38].
Другой гетерозиготный вариант также демонстрировал КДГГ, но без каких-либо других отличительных признаков, и фенотип был не полностью пенетрантным [39].
LHX4 (OMIM 601007)
Мутации характеризуются аутосомно-доминантным или аутосомно-рецессивным наследованием с неполной пенетрантностью. Фенотип включает дефицит СТГ и ТТГ, примерно у половины таких пациентов наблюдается дефицит АКТГ, редко – дефицит гонадотропных гормонов и крайне редко – ПРЛ. Типичными также являются гипоплазия аденогипофиза, недоразвитое седло. Редкие признаки включают порок развития Киари I и тонкую ножку [4, 40–44]. Есть два описанных примера ИДГР [51]. Описанные пациенты с гомозиготной мутацией p.T126М в гене LHX4 из одной неблизкородственной семьи обладали чертами типичного гетерозиготного пациента LHX4 (КДГГ, аплазия аденогипофиза, дефект седла). Однако помимо типичных проявлений оба брата и сестра обладали следующими фенотипическими признаками: гипоплазией средней части лица, маленьким вздернутым носом с вдавленной переносицей, низко посаженными ушами; все погибли в течение первой недели жизни, предположительно от молниеносного сепсиса [45].
FOXA2 (OMIM 600288)
FOXA2 – ген ядерного фактора гепатоцитов 3β, расположенный на 20-й хромосоме. К настоящему времени описано несколько пациентов с гетерозиготными вариантами FOXA2. Они характеризуются признаками МДГГ (СТГ, ТТГ, АКТГ) с высокой распространенностью гиперинсулинемии, гипоплазией/аплазией аденогипофиза и рядом особенностей, таких как единственный центральный резец, дисморфические черты лица, аномалии желчевыводящих путей, пороки сердца и задержка развития нервной системы [46–48]. Генотип чаще всего характеризуется микроделецией или делецией, затрагивающими 20p11.21. FOXA2 и наследуемыми аутосомно-доминантно [5].
NFKB2 (OMIM 164012)
Ген, кодирующий субъединицу комплекса ядерного фактора транскрипции – каппа-B (NFkB). Комплекс NFkB экспрессируется во многих клетках человека и действует как центральный активатор генов, участвующих в воспалении и иммунной функции. Наследуется аутосомно-доминантно, фенотип заболевания состоит из центральной надпочечниковой недостаточности и вариабельного иммунодефицита, куда реже встречается СТГ-дефицит. На МРТ головного мозга часто можно обнаружить гипоплазию аденогипофиза [49].
Есть описание семьи, в которой у одного сибса развилась клиника иммунодефицита, дефицита АКТГ, СТГ и ТТГ, у второго – только иммунодефицита, что говорит о неполной пенетрантности гена [48, 50].
PROKR2 (OMIM 607123)
Ген, кодирующий G-белок-связанный прокинетициновый рецептор 2-го типа, открытый в 2006 г. C. Dodé et al. [53]. В отличие от рецептора 1-го типа, PROKR2 локализуется преимущественно в центральной нервной системе [51, 52]. Чаще всего мутации в гене PROKR2 с потерей функции этого гена ассоциируются с развитием гипогонадотропного гипогонадизма с аносмией, являясь причиной развития синдрома Кальмана примерно в 9% [53–55]. Но на данный момент уже известно, что фенотип заболевания куда более разнообразный – от изолированного гипогонадотропного гипогонадизма с септооптической дисплазией до пангипопитуитаризма без септооптической дисплазии и с нормальным функционированием обонятельного аппарата [56]. Роль PROKR2 в этиологии развития СОД и гипопитуитаризма неясна, отсутствует четкая корреляция генотип–фенотип. При этом есть описание случаев, когда гомозиготный по мутации родитель был абсолютно фенотипически здоровым, при это у ребенка с аналогичной мутацией развилась СОД.
В 2013 г. M. McCabe et al. проанализировали данные 422 пациентов с гипопитуитаризмом и СОД (375/89%) или голопрозэнцефалией (47/11%). У 11 (2,6%) неродственных пациентов ученые обнаружили мутации в кодирующей области гена PROKR2, при этом 9 из вариантов уже были описаны и ассоциировались ранее с СК. Фенотип пациентов включал развитие гипопитуитаризма – от ИДГР, сочетания дефицита СТГ и ТТГ до пангипопитуитаризма с развитием у одного из них несахарного диабета. По данным МРТ головного мозга обнаружили разброс изменений от изолированной гипо-/аплазии аденогипофиза до сочетания аплазии аденогипофиза с дисгенезией мозолистого тела, мальформацией Денди–Уокера и гипоплазией мозжечка [57].
FGF8 (OMIM 600483)/ FGFR1 (OMIM 136350)
Фактор роста фибробластов (FGF) 8 является основным лигандом рецептора FGFR1 и играет решающую роль в формировании передней срединной линии в переднем мозге [58–62]. Сверхэкспрессия FGF8 стимулирует развитие меланотрофов и кортикотрофов, подавляет развитие гонадотрофов, соматотрофов, тиреотрофов и лактотрофов [63]. Гетерозиготные мутации FGFR1 и FGF8 впервые были зарегистрированы в 10% случаев синдрома Кальмана и в 7% нормоосмического гипогонадизма [64]. Пенетрантность была неполной. На МРТ головного мозга регистрируется нормальный или гипоплазированный аденогипофиз и частая встречаемость эктопии задней доли гипофиза. Сообщалось о таких аномалиях, как гипоплазия ушных раковин, агенезия зубов, расщелина неба и дистальные пороки развития конечностей. Мутации FGFR1 и FGF8 также были зарегистрированы у пациентов с септооптической дисплазией, распространенность которой составляет около 4% [65].
PAX6 (OMIM 607108)
PAX6 – регулятор развития глаза, гетерозиготные варианты которого у людей вызывают врожденные аномалии глаз (аниридия, гипоплазия фовеальной области, аномалии зрительного нерва и аномалия Петерса). C. Kioussi et al. выявили, что экспрессия PAX6 необходима для установления четкой границы между дорсальными и вентральными типами клеток передней доли гипофиза, а отсутствие экспрессии PAX6 приводит к заметному увеличению тиреотропной клеточной линии одновременно со значительным снижением соматотропной и лактотропной клеточных линий [66].
На сегодняшний день известно более 350 вариантов PAX6, большинство из них обнаружены в гетерозиготном состоянии. Биаллельные варианты крайне редки, из описанных пациентов трое были мертворожденными или умерли в раннем младенчестве, у всех выявлены аномалии центральной нервной системы. У другого пациента с биаллельным вариантом по данным МРТ головного мозга диагностировали гипоплазию аденогипофиза. Все это подтверждает роль PAX6 в развитии гипофиза у человека [67, 68].
Фенотипический спектр у пациентов с мутациями в гене PAX6 крайне широк – от внешне нормального фенотипа до серьезных, несовместимых с жизнью пороков развития головного мозга и тяжелых патологий глазного дна. Признаки характеризуются неполной пенетрантностью с различными клиническими проявлениями, у членов одной семьи могут ассоциироваться с глазными аномалиями или нет.
L. Hergott-Faure et al. в 2012 г. представили данные детального исследования гипофиза с помощью стимуляционного тестирования пяти носителей мутации PAX6 [69]. В этой работе они не выявили признаков гипопитуитаризма, помимо незначительного дефицита АКТГ. N. Shimo et al. сообщили об одном случае мутации PAX6 с нерезко выраженным гипогонадотропным гипогонадизмом и пограничным дефицитом СТГ. Однако этот конкретный случай неубедителен, поскольку исследуемая пациентка была с ожирением (которое ослабляет реакцию СТГ на стимуляцию СТГ), нормальным ростом 165 см, нормальной менструацией и самостоятельной беременностью [70].
BMP4 (OMIM 112262)
Ген BMP4 расположен на хромосоме 14q22-q23 рядом с вышеописанным геном OTX2, который является хорошо известной причиной развития микрофтальма и анофтальма. Впервые делеция гена BMP4 описана в сочетании с делецией OTX2 у трех пациентов с анофтальмом, дефектами развития гипофиза, задержкой развития и структурными аномалиями головного мозга в сочетании с синдактилией, брахидактилией, аномалиями мочеполовой системы у одного пациента [71, 72]. В 2008 г. опубликован случай изолированного поражения гена BMP4 у пациента с врожденной глаукомой и склерокорнеа, постаксиальной полисиндактилией, аномалиями головного мозга и задержкой развития [73].
В дальнейшем ряд авторов отмечали роль мутации в гене при развитии SHORT-синдрома – редкого заболевания, впервые описанного в 1975 г. и характеризующегося низким ростом, гиперэктензией суставов, паховой грыжей, аномалией Ригера, задержкой прорезывания зубов. Так, L.M. Reis et al. в 2011 г. проанализировали имеющиеся данные о вариантах BMP4 и указали на их ассоциацию с дисгенезией переднего сегмента глаза, плохим ростом и/или массой тела (SDS<-2), макроцефалией, патологией черепно-лицевого развития, что соответствует особенностям SHORT-синдрома. Однако не все пациенты с вариантами BMP4 демонстрируют полный фенотип SHORT-синдрома, что объясняется различной фенотипической экспрессивностью BMP4-мутаций и модификацией их эффекта (эффектов) другими генетическими факторами, расположенными в других частях генома. Наблюдаемая неполная пенетрантность/изменчивая экспрессивность мутаций BMP4 и их широкий фенотипический спектр подтверждают такую возможность [74].
Синдром прерывания ножки гипофиза
Синдром прерывания ножки гипофиза (СПНГ) – редкое явление с частотой встречаемости 0,5/1 000 000 рождений [75]. СПНГ характеризуется истончением или отсутствием ножки гипофиза, гипоплазией или аплазией аденогипофиза и эктопией задней доли гипофиза по данным МРТ [76]. Может ассоциироваться с дефектами средней линии и гипопитуиатризмом – от ИДГР до МДГГ [77]. Прогрессирующее развитие дефицита гормонов приводит к пангипопитуитаризму, но функция нейрогипофиза обычно сохранна, иногда она может быть нарушена в зависимости от его положения. Позже у детей могут наблюдаться низкий рост, снижение скорости роста, судороги, гипотония, интеллектуальная задержка и задержка полового созревания.
ARNT2 (OMIM 606036)
ARNT2 – ген ядерного транслокатора арильного углеводородного рецептора, член нового семейства транскрипционных факторов, картированный у человека в 1997 г. T. Nagase et al. [78]. Недавние исследования показали экспрессию ARNT2 в центральной нервной системе, включая гипоталамус, а также в почках в период эмбриогенеза у человека. В настоящее время имеется описание одной семьи, включающей шестерых детей, рожденных от близкородственного брака, с гомозиготным вариантом со сдвигом рамки считывания гена ARNT2 или с синдромом Уэбба–Даттани. При сборе генеалогического анамнеза выявлены данные о множественных выкидышах в одной ветви семьи. Клинические особенности включали лобно-височную гипоплазию (выступающий лоб, глубоко посаженные глаза, ретрогнатия), выраженную задержку развития, гипофизарную и гипоталамическую недостаточность вследствие гипопластического развития этих областей мозга. Лобные и височные доли также были гипоплазированы, с тонким мозолистым телом и общей задержкой миелинизации головного мозга, особенно в моторной и затылочной коре. У всех детей постнатально отмечался МДГГ (несахарный диабет, вторичные гипотиреоз, гипокортицизм и дефицит гормона роста). В течение первого года жизни развились вторичная микроцефалия, судороги, спастичность.
У 2 из 6 детей исследование глазного дна выявило постретинальную слепоту, также среди клинических проявлений наблюдались аномалии почек (гидронефроз, пузырно-мочеточниковый рефлюкс и нейрогенный мочевой пузырь при сохранной функции почечных клубочков). Трое из шестерых детей умерли до 5 лет [79, 80].
PNPLA6 (OMIM 603197)
Варианты гена PNPLA6, кодирующего целевую эстеразу невропатии, связаны со спектром редких нейродегенеративных заболеваний, включая спастическую параплегию 390-го типа, синдромы Гордона–Холмса и Буше–Нойхаузера. Недавно этот ген связали еще с двумя нейродегенеративными заболеваниями: синдромами Оливера–МакФарлейна и Лоуренса–Муна. Фенотипы этих заболеваний характеризуются хориоретинопатией, спиноцеребеллярной атаксией, спастической параплегией, трудностями в обучении и трихомегалией. Помимо этого они включают дисфункцию гипофиза с гипоплазированным аденогипофизом на МРТ, развитием СТГ-недостаточности и гипогонадотропного гипогонадизма. Исследования экспрессии гена PNPLA6 показали его важную роль в развитии глаз, гипофиза и мозга [81–83].
IFT172 (OMIM 607386)
Ген IFT172 кодирует субъединицу субкомплекса интрафлагеллярного транспорта (IFT-B), необходимого для сборки и поддержания цилиарного аппарата. Варианты IFT172 ранее связывали со скелетными цилиопатиями, с полидактилией или без нее, которые в свою очередь часто ассоциируются с пороками развития сетчатки, мозжечка или гепаторенальной системы [84, 85]. Описан пациент с задержкой роста, возникшей в раннем детском возрасте, гипоплазией аденогипофиза и эктопией нейрогипофиза по данным МРТ, у которого обнаружили сложную гетерозиготную мутацию в IFT172, p.C1727R и новую мутацию сайта сплайсинга в интроне 4 и c.337-2A>C. У этого пациента наблюдалась ретинопатия, ассоциированная с метафизарной дисплазией и гипертонией на фоне почечной недостаточности, что указывает на цилиопатию [86].
ROBO1 (OMIM 602430)
ROBO1 – ген, кодирующий рецептор из семейства рецепторов молекул адгезии нервных клеток. Это еще один ген, который недавно был связан с СПНГ: гетерозиготный вариант со сдвигом рамки считывания, нонсенс и миссенс-варианты (p.A977Qfs*40, p.Y1114* и p.C240S соответственно) были выявлены у пяти пациентов A. Bashamboo et al. При этом родители всех пациентов были фенотипически здоровы. У четырех из пяти пациентов (2 семейных и 1 спорадический случай) наблюдались аномалии глаз, включая гиперметропию с косоглазием и птоз. В 3 случаях был выявлен ИДГР, в 2 других – дефицит СТГ и ТТГ [87]. S. Dateki et al. идентифицировали новый гомозиготный вариант в ROBO1 (c.1342+1G>A) у 5-летнего мальчика. Клиника включала МДГГ, задержку психомоторного развития, тяжелую умственную отсталость, нейросенсорную тугоухость, косоглазие [88]. Z. Liu et al. описали мальчика с МДГГ и СПНГ вследствие миссенс-мутации (c.1690C>T, p.Pro564Ser) в гене ROBO1, унаследованной от матери [89].
IGSF1 (OMIM 300137)
IGSF1 – член суперсемейства иммуноглобулинов-1, гликопротеин плазматической мембраны, кодируемый IGSF1. Ген состоит из 20 экзонов и расположен в районе Xq26. Патогенные варианты IGSF1 вызывают синдром Х-сцепленного дефицита IGSF1, а характерные особенности у пациентов мужского пола включают центральный гипотиреоз, задержку пубертатного повышения тестостерона, скачок роста на фоне нормального или ускоренного роста яичек (дисгармоничное пубертатное развитие) и макроорхидизм у взрослых с относительно низкими концентрациями тестостерона в сыворотке. У части больных мужчин наблюдаются дефицит пролактина, увеличение окружности талии, снижение контроля внимания, низкий рост, а у 16% наблюдается дефицит СТГ. Однако дефицит СТГ носит временный характер, и у взрослых пациентов может наблюдаться повышенная секреция СТГ. Степень и возраст появления центрального гипотиреоза варьируются: у некоторых пациентов она проявляется в младенчестве или детстве, у некоторых пациентов в более старшем возрасте диагностируется легкий гипотиреоз на основании семейных исследований. С учетом редкости этого синдрома полный спектр его фенотипа и патофизиология остаются предметом исследования [90].
Заключение
В данном обзоре мы обобщили основные этиологические факторы и фенотипы гипопитуитаризма, связанного или не связанного с экстрагипофизарными аномалиями. За последние десятилетия понимание механизмов, участвующих в онтогенезе гипофиза, значительно возросло. Со времени первого описания вариантов POU1F1 у человека, ответственных за четко определенный фенотип без внегипофизарных пороков развития, появляется все больше информации о генетических дефектах в факторах транскрипции с различной степенью корреляции фенотип–генотип. К настоящему времени, несмотря на огромный опыт и количество знаний о генетических причинах изолированной или множественной недостаточности гипофиза, этиология большинства (80–90%) врожденных случаев гипопитуитаризма остается невыясненной. Выявление новых причин важно как для постнатальной диагностики для улучшения прогноза и своевременного лечения больных (отсроченная недостаточность гипофиза, дифференциальная диагностика образований гипофиза при МРТ и т.д.), так и в качестве пренатальной диагностики для снижения риска ранней смерти (например, недиагностированный дефицит АКТГ).
Финансирование. Работа проведена в рамках темы госзадания 123021000045–4 «Генетическая персонификация редких вариантов задержки роста и полового развития у детей».
Дополнительная информация
Публикация статьи осуществляется в рамках диссертационной работы Райкиной Е.Н. на соискание ученой степени канд. мед. наук: «Моногенные формы гипопитуитаризма: молекулярно-генетическая гетерогенность и клинический полиморфизм».


