Распространенность сахарного диабета (СД) во всем мире достигла эпидемических уровней и продолжает неуклонно возрастать. По данным Всемирной организации здравоохранения (ВОЗ), в 2000 г. в мире насчитывалось около 160 млн больных СД; эпидемиологи ВОЗ прогнозируют, что к 2025 г. численность больных СД превысит 400 млн человек, из которых 80–90 % составят больные СД 2 типа (СД2) [1].
Рост числа случаев СД сопровождается повышением частоты сосудистых осложнений, которые в свою очередь являются причиной слепоты, хронической почечной недостаточности, ишемической болезни сердца (ИБС), инфаркта миокарда, инсульта, периферической полиневропатии и тяжелых нарушений функционирования центральной нервной системы (ЦНС), приводящих к ранней инвалидизации таких пациентов и высокой летальности. Возраст больных на момент дебюта СД2 постепенно снижается, отмечается тревожное повышение риска развития СД2 у детей и подростков [2]. Раннее развитие СД, особенно у молодых людей, связано с более частой ранней смертностью, различными осложнениями, приводящими к инвалидности, сниженной социальной активности и низкому качеству жизни.
Исследования показали, что на момент постановки диагноза СД2 около 50 % больных уже имеют макро- и микрососудистые осложнения. Возможно, это результат того, что метаболические нарушения возникают гораздо раньше первых клинических проявлений СД. Долгосрочные последствия эпидемии диабета выливаются в огромные человеческие страдания и экономические затраты.
Главным фактором риска развития СД2 является наличие у пациента избыточной массы тела, особенно при отложении жира в абдоминальной области. В последние десятилетия ученые стали рассматривать различные метаболические нарушения или заболевания, ассоциированные с избыточной массой тела, в комплексе, высказываются предположения об общности этих процессов. В 1960-е гг. делались попытки объединения взаимосвязанных метаболических нарушений, ускоряющих развитие макрососудистых атеросклеротических заболеваний и СД2. В 1988 г. американский ученый G. Reaven, объединив нарушения углеводного обмена, артериальную гипертензию (АГ) и дислипидемию в понятие “синдром Х”, впервые высказал предположение о том, что основой этих нарушений могут быть инсулинорезистентность (ИР) и компенсаторная гиперинсулинемия (ГИ) [3]. В конце прошлого века метаболические нарушения и заболевания, развивающиеся у лиц с ожирением, объединили в понятие “метаболический синдром” (МС).
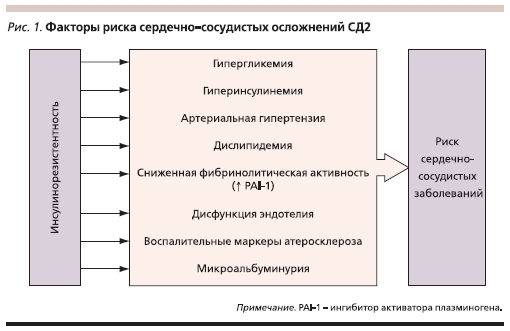
МС – это сочетание метаболических нарушений, в патогенезе которых важную роль играет ИР и которые являются факторами риска раннего развития атеросклероза, его сердечно-сосудистых осложнений (рис. 1). В настоящий момент существует пять групп диагностических критериев МС.
Отечественными учеными, экспертами Всероссийского научного общества кардиологов, также были разработаны и опубликованы критерии МС. Основной критерий – это центральный (абдоминальный) тип ожирения: окружность талии > 80 см у женщин и > 94 см у мужчин.
Дополнительными критериями являются:
• АГ (артериальное давление [АД] ≥ 130/85 мм рт. ст.);
• повышение уровня триглицеридов (≥ 1,7 ммоль/л);
• снижение уровня холестерина липопротеидов высокой плотности (< 1,0 ммоль/л у мужчин и < 1,2 ммоль/л у женщин);
• повышение уровня холестерина липопротеидов низкой плотности > 3,0 ммоль/л);
• гипергликемия натощак (глюкоза в плазме крови ≥ 6,1 ммоль/л;
• нарушение толерантности к глюкозе (глюкоза в плазме крови через 2 часа после нагрузки глюкозой в пределах ≥ 7,8 и ≤ 11,1 ммоль/л.
Наличие у пациента центрального ожирения и двух дополнительных критериев является основанием для диагностирования у него МС.
Клиническая значимость нарушений и заболеваний, объединенных в рамки МС, заключается в том, что их сочетание в значительной степени ускоряет развитие и прогрессирование заболеваний, связанных с атеросклерозом, которые имеют не только медицинское, но и социальное значение в современном обществе. Кроме этого многие исследователи рассматривают МС как прелюдию к СД2: риск развития диабета у лиц с МС в среднем в 5–9 раз выше, чем у лиц без него [4]. По данным статистики, у лиц с впервые диагностированным СД2 уже при первом обращении к врачу выявляются макро- и микрососудистые осложнения этого заболевания: нарушение зрения вследствие диабетической ретинопатии, нарушение функции почек вследствие диабетической нефропатии, поражение сосудов сердца, мозга, сосудов нижних конечностей и др. А при развившемся СД2 риск развития сердечно-сосудистых заболеваний (ССЗ) в 3–4 раза выше, чем без него [5]. Именно эти осложнения являются основной причиной высокой инвалидизации и смертности больных СД2: до 70 % таких пациентов умирают от инфаркта миокарда или инсульта и их последствий.
Механизмы развития ССЗ и СД2 у пациентов с МС: роль ИР, ГИ и постпрандиальной гипергликемии
Ключевым звеном патогенеза МС являются первичная ИР и компенсаторная ГИ. ИР – это нарушение инсулинопосредованной утилизации глюкозы клетками.
Различают несколько видов ИР, сопровождающих ряд физиологических и патологических процессов:
• физиологическая ИР выявляется в пубертатном периоде, при беременности, в климактерическом периоде, во время ночного сна, при богатой жиром диете;
• метаболическая ИР характерна для МС, СД2, декомпенсированного СД1, диабетического кетоацидоза, ожирения, выраженной недостаточности питания, гиперурикемии, гипогликемии, индуцированной инсулином, злоупотреблением алкоголем;
• эндокринная ИР отмечается при тиреотоксикозе, гипотиреозе, синдроме Кушинга, акромегалии, феохромоцитоме;
• неэндокринная ИР типична для АГ, хронической почечной недостаточности, цирроза печени, сердечной недостаточности, ревматоидного артрита, черного акантоза, миотонической дистрофии, травм, ожогов, сепсиса, состояния после хирургических вмешательств, раковой кахексии.
Наибольшее клиническое значение имеет потеря чувствительности к инсулину мышечной, жировой и печеночной тканей. Предполагают, что причиной ускоренного атерогенеза и высокой летальности от ИБС и инсультов у больных СД2 также могут быть ИР и сопутствующая ей ГИ. У пациентов с ИР имеются дефекты генов, ответственных за передачу сигнала после соединения инсулина со своим рецептором
(пострецепторные дефекты). Прежде всего у них нарушаются транслокация и синтез внутриклеточного транспортера глюкозы ГЛЮТ-4. Генетические дефекты могут находиться на уровне субстрата рецептора инсулина типа 1 и/или фосфатидилинозитол-3-киназы. Обнаружено также нарушение экспрессии и других генов, обеспечивающих метаболизм глюкозы и липидов, таких как глюкозо-6-фосфатдегидрогеназа, глюкокиназа, липопротеинлипаза, синтаза жирных кислот и др.
В отсутствие необходимых для этого внешних факторов (избыточного калорийного питания, особенно жирной пищи, и низкой физической активности) генетическая предрасположенность к ИР может не реализоваться и не проявиться клинически (в виде МС и/или СД2). Эти факторы сами по себе способствуют увеличению абдоминального ожирения, накоплению свободных жирных кислот (СЖК) и, следовательно, усилению имеющейся ИР. Развивающаяся при ИР компенсаторная ГИ, с одной стороны, позволяет вначале поддерживать углеводный обмен в норме, с другой – способствует развитию метаболических, гемодинамических и органных нарушений, в конечном итоге приводящих к развитию ССЗ и СД2.
ИР мышечной ткани проявляется в снижении поступления глюкозы из крови в миоциты и ее утилизации в мышечных клетках. При ИР жировой ткани наблюдается резистентность к антилиполитическому действию инсулина, что приводит к накоплению СЖК и глицерина. Поступающие в печень СЖК становятся основным источником формирования атерогенных липопротеидов очень низкой плотности. В ткани печени ИР характеризуется снижением синтеза гликогена и активацией процессов распада гликогена до глюкозы (гликогенолиз) и синтеза глюкозы de novo из аминокислот, лактата, пирувата, глицерина (глюконеогенез), в результате чего глюкоза из печени поступает в кровоток.
В целом ИР является эволюционно закрепленным механизмом выживания в неблагоприятных условиях, когда периоды изобилия чередовались с периодами голода. Наличие ИР обеспечивало накопление энергии в виде отложений жира, запасов которого хватало на то, чтобы пережить голод [6]. В современных условиях в странах с высоким экономическим развитием, сопутствующим изобилием и склонностью к малоподвижному образу жизни сохранившиеся в генетической памяти механизмы ИР продолжают “работать” на накопление энергии, что способствует развитию абдоминального ожирения, дислипидемии, раннему атеросклерозу, АГ и СД2.
К настоящему времени опубликованы результаты более 10 клинических исследований с участием не менее 15 тыс. человек, которые позволяют утверждать, что ИР и сопутствующая ей ГИ являются факторами риска ускоренного атерогенеза и высокой летальности от ИБС. Кроме того, имеются
клинические доказательства того, что ГИ является независимым фактором риска развития ИБС у лиц без СД2 (по данным исследований: Paris prospective Study – около 7 тыс. обследованных, Busselton – более 1 тыс. обследованных и Helsinki Policemen Study – 982 обследованных) [7]. В последние годы это подтверждено и у больных СД2. Этим данным есть экспериментальное обоснование. Работы R. Stout свидетельствуют о том, что инсулин оказывает прямое атерогенное действие на стенки сосудов, вызывая пролиферацию и миграцию гладкомышечных клеток, синтез в них липидов, пролиферацию фибробластов. Таким образом, ИР и ГИ вносят весомый вклад в прогрессирование атеросклероза как у лиц без СД, так и у больных СД2 [8].
Существенную роль играет ИР в развитии АГ. Взаимосвязь ГИ (маркера ИР) и эссенциальной АГ настолько прочна, что при высокой концентрации у больного инсулина плазмы у него в скором времени можно прогнозировать развитие АГ. Причем эта связь прослеживается у лиц как с ожирением, так и с нормальной массой тела. Существует несколько механизмов, объясняющих повышение АД при ГИ (рис. 2). Инсулин способствует активации симпатической нервной системы (СНС), повышению реабсорбции Na и воды в почечных канальцах, внутриклеточному накоплению Na и Ca. Инсулин как митогенный фактор активирует пролиферацию гладкомышечных клеток сосудов, что ведет к утолщению их стенки.
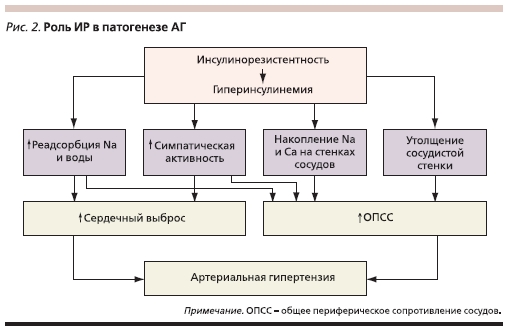
Механизм влияния инсулина на СНС до конца не ясен. Предполагают, что инсулин может активировать СНС путем прямого воздействия на ЦНС, проникая через гематоэнцефалический барьер в перивентрикулярную область гипоталамуса, где, связываясь со своими рецепторами на поверхности нейронов, блокирует активность парасимпатической нервной системы и, напротив, активирует СНС. G.M. Reavean – основоположник синдрома ИР – предположил, что причиной гиперактивации СНС в условиях гипергликемии может быть повышенный метаболизм глюкозы в ядрах гипоталамуса, что тормозит передачу блокирующих импульсов на симпатические центры продолговатого мозга. Стимуляция СНС при ГИ сопровождается увеличением сердечного выброса, повышением общего периферического сосудистого сопротивления, что неизбежно приводит к повышению АД. Одновременное снижение активности парасимпатической системы, вызванное ГИ, увеличивает частоту сердечных сокращений.
Усиление реабсорбции Na и воды также происходит под влиянием ГИ. Инсулин оказывает прямое воздействие на проксимальные канальцы нефронов, повышая реабсорбцию Na и жидкости. Помимо антинатрийуреза инсулин вызывает антикалийурез и антиурикозурию. В результате увеличивается объем циркулирующей жидкости, что приводит к повышению сердечного выброса. Внутриклеточное накопление Na и Ca – эффект действия инсулина. Инсулин блокирует активность Na/К- и Ca/Mg-АТФазы клеточных мембран, что приводит к повышению внутриклеточного содержания Na и Ca. Вследствие накопления этих электролитов в стенке сосудов повышается чувствительность сосудистых
рецепторов к действию сосудосуживающих факторов.
Под влиянием инсулина происходит утолщение стенки сосудов. Митогенные свойства инсулина обнаружены достаточно давно в серии экспериментальных работ, где было показано, что инсулин стимулирует клеточный рост, пролиферацию и миграцию гладкомышечных клеток сосудов, приводит к утолщению их стенки. В норме инсулин, связываясь с рецепторами на поверхности клеток эндотелия, может действовать двумя различными путями. Первый путь – это активация секреции оксида азота (NO) через субстраты инсулиновых рецепторов-1 и -2 (IRS-1 и IRS-2) и фосфатидилинозитол-3-киназу (PI3-K). Этот механизм обеспечивает сосудорасширяющие и антиатерогенные свойства инсулина, участвует в инсулинозависимом транспорте глюкозы в клетки. Второй путь – реализация митогенных свойств инсулина через каскад посредников, повышающих активность митогенактивированной протеинкиназы (МАРК), что завершается пролиферацией и миграцией гладкомышечных клеток, активацией синтеза сосудосуживающего фактора эндотелина-1 и повышением АД. Оказалось, что в условиях ИР первый механизм не работает – именно этот путь резистентен к действию инсулина, следовательно, молекула NO не синтезируется. В то же время второй механизм сохраняет свою высокую активность. Поэтому ГИ, развивающаяся вследствие ИР (при метаболическом синдроме, СД2, висцеральном ожирении), не только не снижает АД, а напротив, оказывает гипертензивное и атерогенное действия.
Существует взаимосвязь между активностью ренин-ангиотензиновой системы (РАС), уровнем АД и чувствительностью тканей к инсулину. Хорошо известно, что гиперактивность РАС стойко поддерживает высокое АД. Однако лишь недавно в экспериментальных условиях получены убедительные данные о том, что ангиотензин II (АТII) дозозависимо ингибирует пострецепторную сигнальную систему инсулина, реализующую транспорт глюкозы в клетки и продукцию NO [9]. Одновременно АТII стимулирует МАРК, задействованную в осуществлении митогенной и пролиферативной активности инсулина.
Таким образом, гиперактивность РАС и АТII вызывает резистентность тканей к антиатерогенному и антигипертензивному действиям инсулина; это приводит к развитию ССЗ, а также блокирует транспорт глюкозы в клетки, что способствует развитию предиабета, а затем и СД2.
Возможность замедления прогрессирования и уменьшения развития осложнений СД2
Одной из основных причин развития СД2 и его сердечно-сосудистых осложнений является сниженная чувствительность мышечной и жировой тканей, а также печени к действию эндогенного инсулина – ИР. Когда у пациента развивается клиническая картина СД2 – это значит, что ИР тканей, которая существовала у него задолго до дебюта заболевания, уже привела к тому, что эндогенных запасов инсулина перестало хватать на преодоление существующей ИР. Функциональная активность β-клеток поджелудочной железы уже снизилась к тому времени на 50 %, что и привело к повышению уровня гликемии. Эти изменения на стадии клинических проявлений СД безусловно необратимы, поэтому даже раннее начало лечения СД2 не может приводить к полному выздоровлению, но применение лекарственных средств, влияющих на ослабление основных патогенетических влияний, в основном ИР, необходимо на любом этапе развития углеводных нарушений.
Рекомендации немедикаментозного плана включают изменения образа жизни, сочетающие соблюдение диеты и регулярные физические нагрузки, которые должны направляться на снижение массы тела, уменьшение ИР, нормализацию АД, липидных нарушений и восстановление углеводного
обмена.
В случае низкой приверженности пациентов изменению образа жизни, а также при неэффективности немедикаментозных методов коррекции оправданно применение препаратов, повышающих чувствительность тканей к инсулину, т. е. облегчающих поступление глюкозы в голодающую клетку без дополнительной ГИ, следовательно, прерывающих патогенетические связи развития СД и его осложнений. К таким препаратам в первую очередь относятся бигуаниды и тиазолидиндионы (ТЗД; глитазоны), эффективность которых в плане снижения риска развития СД2 и ССЗ у пациентов с СД2 доказана в крупных международных плацебо-контролируемых исследованиях.
Глитазоны
С конца 1950-х гг. фармацевтическая промышленность всего мира работает над созданием препаратов, устраняющих ИР. Препараты группы бигуанидов (метформин) лишь отчасти решают поставленную задачу, устраняя ИР ткани печени, что проявляется снижением выброса глюкозы печенью. В меньшей степени эти препараты влияют на ИР на уровне мышечной и жировой тканей. Только в конце 1990-х гг. появилась принципиально новая группа препаратов – ТЗД, или глитазоны.
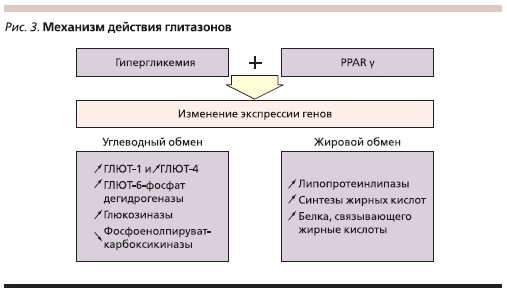
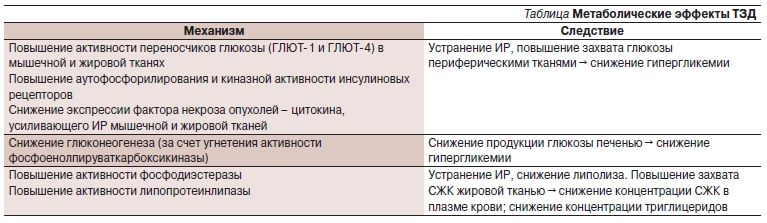
Лекарственные средства этой группы являются синтетическими лигандами рецепторов γ, активируемых пролифератором пероксисом (PPARγ – Peroxisome proliferator-activated receptor gamma). Эти рецепторы располагаются преимущественно в ядрах клеток жировой и мышечной тканей. Их также можно обнаружить в ядрах клеток сердечной мышцы, печени и почек. Соединившись с рецепторами PPARγ в ядрах клеток, ТЗД изменяют транскрипцию генов, регулирующих метаболизм глюкозы и липидов, что в присутствии эндогенного инсулина ведет к повышению транспорта глюкозы и свободных жирных кислот через стенку сосуда в ткань (рис. 3). В результате описанного механизма действия ТЗД вызывают следующие изменения метаболизма углеводов и жиров (см. таблицу).
Таблица. Метаболические эффекты ТЗД.
Иными словами, устраняя ИР, ТЗД усиливают физиологическое действие собственного эндогенного инсулина, при этом снижают его концентрацию в крови.
С момента их открытия было зарегистрировано три препарата:
• троглитазон (Резулин);
• росиглитазон (Авандия);
• пиоглитазон (Актос).
Все три препарата имели сходную структуру, содержащую тиазолидин2-4-дионы. Различия касались лишь боковой цепи, которая определяет фармакологическую активность препаратов. Боковая цепь троглитазона была представлена α-токоферолом. Такая структура предложена для усиления антиоксидантных свойств препарата. Однако за 2 года применения троглитазона (с 1997 по 1999 г.) было зарегистрировано 43 случая развития острой печеночной недостаточности (из них 28 – со смертельным исходом). Высокая гепатоксичность троглитазона послужила поводом для запрещения этого препарата к применению и снятия его с производства в марте 2000 г. При анализе побочных эффектов троглитазона установлено, что осложнения со стороны печени непосредственно связаны с боковой цепью (α-токоферол), входящей в структуру препарата. Два других препарата (росиглитазон и пиоглитазон) не имели в своей структуре указанного соединения. За весь период их применения (с 1999 г. по настоящее время) зарегистрировано лишь 2 случая острой гепатоксичности на фоне лечения росиглитазоном и ни одного – на фоне лечения пиоглитазоном.
Однако в сентябре 2010 г. группу ТЗД покинул еще один представитель: Европейское медицинское агентство (EMA) рекомендовало приостановить в Европе продажу препаратов для лечения СД, содержащих росиглитазон, – Авандию, Авандамет, Авандаглим. Решение о порядке его вывода стало
кульминацией длительного периода сомнений в побочных эффектах росиглитазона, озабоченность которыми росла как снежный ком после опубликованных в мае 2007 г. результатов мета-анализа S. Nissen и соавт. “Влияние росиглитазона на риск развития инфаркта миокарда и смерти от сердечно-сосудистых причин”. Представленные данные всколыхнули медицинскую общественность и вызвали подъем научного интереса к рассматриваемой проблеме.
В своей работе E. Nissen и соавт. [10] проанализировали 42 исследования, соответствовавших следующим критериям включения:
• продолжительность более 24 недель;
• наличие контрольной группы лиц, не получавших росиглитазон;
• регистрация за время испытания по крайней мере одного инфаркта миокарда или смерти от сердечно-сосудистых причин.
В мета-анализ вошли и такие крупные исследования, как ADOPT, DREAM. Все пациенты, принимавшие росиглитазон, были объединены в одну группу, контрольная же группа была представлена больными, получавшими любой другой сахароснижающий препарат или плацебо. Данные мета-анализа показали, что применение росиглитазона ассоциируется с достоверным увеличением риска развития инфаркта миокарда (коэффициент вероятности – 1,43; 95 % доверительный интервал [ДИ] – 1,03–1,98; р = 0,03) и риска смерти от сердечно-сосудистых причин (коэффициент вероятности – 1,64; 95 % ДИ – 0,98–2,74), не достигшего, однако, уровня статистической значимости (р = 0,06). Решение EMA в сентябре 2010 г. было принято после нескольких лет анализа и споров в отношении сердечно-сосудистой безопасности
росиглитазона и в связи с доказанными неблагоприятными эффектами со стороны сердечно-сосудистой системы, в т. ч. возможной связью между приемом росиглитазона и повышением риска развития ИБС. Пациентам, принимавшим росиглитазон, настоятельно было рекомендовано обратиться к лечащему врачу для изменения дальнейшей терапии. В то же время FDA (Food and Drug Administration) приняла решение об оставлении препаратов, содержащих росиглитазон, но настоятельно рекомендовала ограничить контингент пациентов, которые могут их получать.
Для объективной оценки произошедшего необходимо отметить, что параллельно с анализом результатов исследований, в которых использовался росиглитазон, также были оценены и многократно изучены данные по безопасности применения пиоглитазона. Было выявлено, что при одинаковой длительности применения в клинической практике пиоглитазона и росиглитазона (около 10 лет), практически сравнимом числе пациентов, принимавших за то время данные препараты, результаты их воздействия не одинаковы. Пиоглитазон ассоциируется с достоверно меньшим риском смерти, инфаркта миокарда и инсульта в разных группах больных диабетом, т. е. за время терапии пиоглитазоном риск развития сердечно-сосудистых осложнений и смерти снижается. Величина и направленность благоприятного действия пиоглитазона на ишемические события были гомогенны в испытаниях различной продолжительности и одинаковы для пациентов как с установленным заболеванием сосудов, так и без него [11].
Приведем результаты недавно завершенного крупного многоцентрового двойного слепого рандомизированного плацебо-контролируемого исследования PROactive (Prospective Pioglitazone Clinical Trial In Macrovascular Events) [12]. В исследовании приняли участие 5238 пациентов с СД2 и ССЗ, получавших пиоглитазон или плацебо в дополнение к их исходной сахароснижающей терапии. Пациенты, имевшие в анамнезе инфаркт миокарда (n = 2445), были дополнительно объединены в подгруппу, для которой исследователи предварительно задали и определили несколько комбинированных конечных точек. По результатам исследования не зарегистрирована достоверная разница в скорости развития кардиоваскулярных событий или смерти (первичные конечные точки) между двумя группами. Однако при анализе дополнительных точек в обозначенной подгруппе выявлено достоверное снижение риска повторного инфаркта миокарда (5,3 по сравнению с 7,2 %; р = 0,0453) и в целом острого коронарного синдрома (уровень риска – 0,63; р = 0,035) среди пациентов, получавших терапию пиоглитазоном [13]. Такое разное влияние на сердечно-сосудистый риск росиглитазона и пиоглитазона, возможно, объясняется самим механизмом действия этих препаратов – влиянием на генетический потенциал клетки, в результате которого число активируемых и подавляемых генов при использовании препаратов группы ТЗД различно (рис. 4) [14].
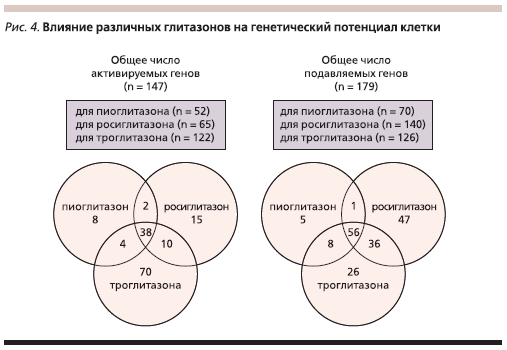
По данным клинических исследований, пиоглитазон демонстрирует способность отдалять развитие СД и замедлять его прогрессирование до формирования сердечно-сосудистых осложнений благодаря уменьшению ИР, снижению секреции инсулина, протекции β-клеток поджелудочной железы, благотворному влиянию на липидный обмен (в отличие от росиглитазона), при этом наблюдается повышение уровня липопротеидов высокой плотности, снижение концентрации триглицеридов, индекса атерогенности [15, 16]. Таким образом, единственным эффективным и безопасным препаратом из группы глитазонов на сегодняшний день остался только пиоглитазон.
Результаты проведенных многочисленных клинических исследований и мета-анализов заставляют сегодня иначе взглянуть на препараты рассматриваемой группы. Высокий риск развития инфаркта миокарда, сердечной недостаточности, нарушений липидного обмена, остеопороза должен заставить практического врача задуматься над соотношением риска и пользы при назначения росиглитазона. В более выгодном свете на этом фоне предстает пиоглитазон с потенциальным снижением риска повторных инфарктов миокарда, положительным влиянием на липидный обмен, осуществляющий профилактику СД2, коррекцию нарушений при неалкогольном стеатогепатозе [17].
В этом году на российский рынок выходит новый препарат, Амальвия (пиоглитазон, Тева), – современное средство лечения СД2 как в виде монотерапии, так и в комбинации с другими пероральными сахароснижающими препаратами и/или инсулином. Пиоглитазон пользуется большой популярностью у врачей всего мира в терапии СД2. По данным сайта www.drugs.com, число выписанных рецептов на пиоглитазон за 2008–2009 гг.
намного опережает число выписанных препаратов сульфонилмочевины. Преимуществами терапии препаратом Амальвия больных СД2 являются улучшение функции β-клеток поджелудочной железы, отсутствие риска гипогликемических состояний, удобство применения (1 раз в сутки вне зависимости от приема пищи). Важным преимуществом применения Амальвии является отсутствие необходимости коррекции дозы для пациентов с нарушением функции почек. Данный факт позволяет рассматривать Амальвию как препарат выбора для пациентов с выраженной ИР и хронической болезнью почек III–V стадий (скорость клубочковой фильтрации < 60 мл/мин), что является противопоказанием к назначению метформина (повышенный риск лактоацидоза).
Таким образом, Амальвию можно применять в качестве монотерапии в дебюте СД2 у пациентов с противопоказаниями к применению метформина, она также может быть альтернативой для больных, получавших росиглитазон; в составе двойной и тройной комбинированной терапии Амальвию назначают с метформином, препаратами сульфонилмочевины или ингибиторами дипептидилпептидазы-4, а также с инсулином. На сегодняшний день в России Амальвия является единственным пиоглитазоном, производимым в Европе в соответствии с международными стандартами, что гарантирует высокое качество и безопасность препарата.

{kind=link}