Рост распространенности сахарного диабета типа 2 (СД2) в мире приобрел характер “неинфекционной эпидемии”, и по прогнозам экспертов ВОЗ, число больных СД2 к 2025 г. должно достичь 380 млн человек [1]. Прогрессирование при СД макро- (поражение коронарных, церебральных и периферических сосудов) и микроваскулярных (диабетическая ретинопатия, нефро- и нейропатия) осложнений выводит его на 3-е место по причинам летальности после сердечно-сосудистых и онкологических заболеваний. Смертность от сердечно-сосудистых болезней лиц с СД2 в 3 раза выше, чем у населения в целом [2]. При этом в 80 % случаев причиной смерти являются атеросклеротические макроваскулярные осложнения [3]. В целом от заболеваний, обусловленных атеросклерозом, умирает больше больных СД, чем от всех других причин вместе взятых [4]. Причиной такого выраженного поражения сосудистого русла в настоящее время считают гипергликемию. Метаанализ 20 различных исследований, включивших 95 783 пациента, наблюдаемых в течение 12 лет, позволил сделать вывод о том, что глюкоза является таким же фактором риска для развития атеросклероза и острой сердечно-сосудистой летальности, как и уровень общего холестерина, а также артериального давления [5].
Ставшие классикой результаты исследования UKPDS [6] и Kumamoto [7], показавшие роль гликемического контроля в снижении риска прогрессирования диабетических сосудистых осложнений, подтвержденные серией недавних крупных рандомизированных исследований, таких как ADOPT [8], ADVANCE [9], PROACTIVE [10], в комбинации с недавно опубликованными данными длительного (10-летнего) наблюдения за пациентами, участвовавшими в исследованиях DCCT/EDIC [11], UKPDS [12] и Steno-2 study [13], послужили основанием для создания международного консенсуса по лечению СД2, в котором уровень гликированного гемоглобина (HbA1c) < 6,5 % признан в качестве конечной цели в лечении этого заболевания.
Современные рекомендации по более агрессивному подходу к достижению целевого уровня гликемического контроля были, тем не менее, пересмотрены в связи с увеличением риска острой сердечно-сосудистой летальности при попытке добиться целевого уровня HbA1c < 6,5 % при проведении исследований RECORD [14], ACCORD [15] и VADT [16].
Таким образом, вопрос об эффективности и безопасности гликемического контроля по сей день остается спорным. Более 35 лет назад были опубликованы результаты Университетской группы по изучению диабета (UGDP), которые показали возрастание сердечно-сосудистой летальности у больных СД2, получавших толбутамид – препарат сульфонилмочевины (ПСМ) I поколения, по сравнению с пациентами, получавшими инсулинотерапию либо плацебо [17]. Результаты вызвали оживленную дискуссию и были поставлены под сомнение, поскольку сама методология исследования не выдерживала критики: не было строгой рандомизации, не отслеживалась приверженность пациентов лечению, в исследование включались также больные, не страдавшие СД2 [18, 19]. Вскоре появились публикации, показавшие идентичные результаты: меньшую выживаемость после инфаркта миокарда (ИМ) у пациентов, получавших сахароснижающую пероральную терапию по сравнению с
диетой [20, 21] или инсулинотерапией [22]. Напротив, другие исследования не выявили какой-либо связи между типом антидиабетического лечения и выживаемостью после ИМ [23], в т. ч. при длительном наблюдении [24], или показали весьма значительное преимущество с точки зрения общей смертности у пациентов, получавших ПСМ, по сравнению с диетой или инсулинотерапией. Однако это были ретроспективные исследования с участием самых различных групп пациентов, которые невозможно адекватно оценить без статистической обработки всех основных противоречивых факторов.
диетой [20, 21] или инсулинотерапией [22]. Напротив, другие исследования не выявили какой-либо связи между типом антидиабетического лечения и выживаемостью после ИМ [23], в т. ч. при длительном наблюдении [24], или показали весьма значительное преимущество с точки зрения общей смертности у пациентов, получавших ПСМ, по сравнению с диетой или инсулинотерапией. Однако это были ретроспективные исследования с участием самых различных групп пациентов, которые невозможно адекватно оценить без статистической обработки всех основных противоречивых факторов.
Этот вопрос оказался закрытым только после публикации результатов UKPDS [6], показавших отсутствие какой-либо заметной разницы в отношениисердечно-сосудистой заболеваемости и смертности между группами больных, получавших лечение инсулином, глибенкламидом (ГБ) или хлорпропамидом, примерно через 10 лет лечения. Открытие гетерогенности рецепторов к ПСМ (SUR), обнаружение SUR2A и SUR2B в кардиомиоцитах и гладкомышечных клетках сосудистой стенки, а также данные о неоднородности взаимодействия различных ПСМ с этими рецепторами в сердце и их влияния на сосудистое русло в экспериментальных моделях, вновь сделали актуальным обсуждение
вопроса о потенциальной кардиотоксичности ПСМ или некоторых из них. В последнее время появилось огромное количество публикаций на эту тему с разными выводами: некоторые предлагают запретить потенциально опасные ПСМ у больных СД, страдающих коронарной болезнью сердца [25, 26], тогда как в других указывается на отсутствие достаточных оснований для исключения этих препаратов из арсенала сахароснижающих средств.
вопроса о потенциальной кардиотоксичности ПСМ или некоторых из них. В последнее время появилось огромное количество публикаций на эту тему с разными выводами: некоторые предлагают запретить потенциально опасные ПСМ у больных СД, страдающих коронарной болезнью сердца [25, 26], тогда как в других указывается на отсутствие достаточных оснований для исключения этих препаратов из арсенала сахароснижающих средств.
Механизм действия ПСМ
Хорошо известно, что ПСМ могут оказывать сахароснижающий эффект только при сохраненной способности инсулярного аппарата к секреции гормона. ПСМ оказывают стимулирующий эффект на секрецию инсулина за счет связывания со специфическими рецепторами плазматической мембраны β-клетки, интегрированными в структуру АТФ-зависимых К+-каналов плазматических мембран [27]. Роль КАТФ-каналов в процессе регуляции инсулиновой секреции является ключевой. При поступлении в β-клетку глюкозы и при ее окислении повышается концентрация АТФ, что сопровождается закрытием КАТФ-каналов, которое ведет к изменению мембранного потенциала. Деполяризация мембраны сопровождается открытием вольтаж-зависимых Са++-каналов и вхождением ионов Са++ в клетку. В результате повышается концентрация внутриклеточного Са++, являющегося сократительным микроэлементом, благодаря чему происходит сокращение внутриклеточных миофибрилл и стимулируется секреция инсулина путем экзоцитоза. При взаимодействии ПСМ с рецепторами β-клетки происходит закрытие КАТФ-каналов и инициируется вся цепь вышеописанных событий, заканчивающаяся секрецией синтезированного ранее и накопленного в β-клетке инсулина (рис. 1). Благодаря тому что ПСМ повышают чувствительность β-клеток к глюкозозависимому инсулинотропному полипептиду (ГИП), стимуляция секреции инсулина происходит в соответствии с уровнем глюкозы, вследствие чего восстанавливается нормальная кривая инсулиновой секреции, в частности 1-я фаза, необходимая для снижения постпрандиальной гликемии.
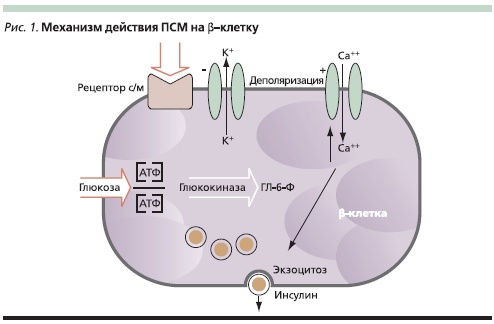
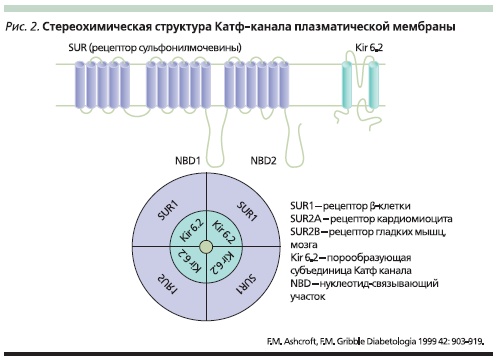
Чтобы ответить на вопрос, почему различные ПСМ имеют различную сахароснижающую активность, рассмотрим более детально структуру КАТФ-каналов и сопоставим ее с химической структурой ПСМ. Результаты современных исследований по изучению регуляции секреции инсулина [27] представляют КАТФ-канал как композицию двух типов субъединиц – порообразующей Kir6.x и регуляторной, способной связывать СМ и потому названной рецептором СМ – SUR. Эти субъединицы стереохимически объединены в соотношении 4 : 4, образуя октаометрический комплекс из четырех пороформирующих субъединиц (Kir6.2) и четырех регуляторных SUR-субъединиц (рис. 2). Описано два гена, кодирующих Kir6.1 и Kir6.2, равно как и два гена, кодирующих SUR1 и SUR2. Электрофизиологические исследования показали, что различные комбинации Kir.х и SUR присутствуют в разных тканях. Kir6.2 экспрессируются в β-клетках, сердце, мозге и скелетной мускулатуре, а Kir6.1 формируют пору КАТФ-канала в гладкой мускулатуре, хотя существуют данные, что здесь могут присутствовать и Kir6.2-, и Kir6.1-субъединицы, формируя пору КАТФ-канала с различными свойствами. SUR1 образуют регулирующую субъединиицу в β-клетках и некоторых видах нейронов, SUR2A – в сердечной и скелетной мускулатуре, SUR2B – в гладкой мускулатуре. Порообразующая Kir6.2-субъединица состоит из двух трансмембранных доменов, связанных между собой, а регулирующая SUR-субъединица – из 17 трансмембранных доменов, объединенных в три группы, и двух внутриклеточных нуклеотид-связывающих доменов (рис. 2). Поскольку до сих пор не найдено АТФ-связывающих участков на Kir6.2-субъединице, считается, что именно SUR-субъединица, в частности ее нуклеотид-связывающие домены, ответственна за метаболическую регуляцию КАТФ-канала. Именно благодаря связыванию аденозина с нуклеотид-связывающим доменом SUR при повышении концентрации АТФ в клетке происходит инактивация КАТФ-канала и пора закрывается, что обусловливает деполяризацию мембраны. И напротив, при снижении концентрации АТФ происходят открытие КАТФ-канала и реполяризация мембраны.
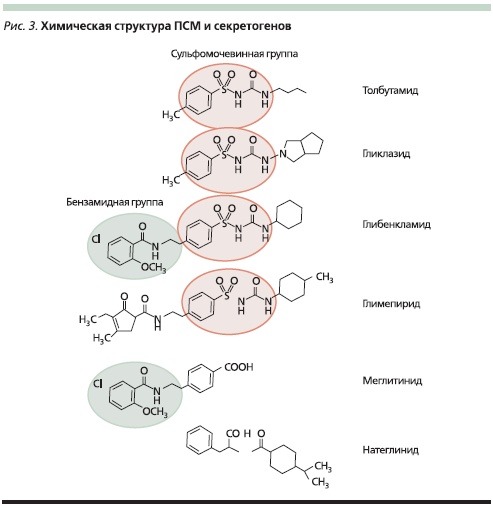
Каким же образом ПСМ ингибируют КАТФ-канал? Как показали исследования последних лет, SUR1-субъединица КАТФ-канала β-клетки связывает с высокой константой сродства ПСМ, однако эта константа различна для разных представителей этой группы. Самой слабой константой обладает препарат I поколения – толбутамид, самой высокой – ГБ; по-видимому, этим фактом и объясняются различия в сахароснижающей активности препаратов, поскольку, чем выше сродство ПСМ к рецептору, тем длительнее его ингибирующее влияние на КАТФ-канал и тем сильнее будет
стимулироваться секреция инсулина за счет поступления в β-клетки ионов Са++. Вместе с тем показано, что помимо СМ SUR1 связывает еще и производные бензойной кислоты, а также фенилаланина (меглитиниды), в своей структуре содержащие т. н. бензамидную группировку (рис. 3). Последняя связывается с SUR c более низкой константой сродства, вот почему новая группа сахароснижающих препаратов, т. н. секретогенов, стимулирует секрецию инсулина значительно слабее и эффект их действия заканчивается быстрее. С другой стороны, с учетом особенностей химической структуры ГБ, содержащего и сульфонилмочевинную, и бензамидную группировки, становится ясно, почему до сего времени он остается “золотым стандартом”, с которым сравнивают активность любого нового сахароснижающего препарата: соединяясь с двумя связывающими
местами на SUR1, ГБ наиболее быстро и мощно способствует закрытию КАТФ-канала, стимулируя деполяризацию мембраны β-клетки, повышение внутриклеточного Са++ и секрецию инсулина.
стимулироваться секреция инсулина за счет поступления в β-клетки ионов Са++. Вместе с тем показано, что помимо СМ SUR1 связывает еще и производные бензойной кислоты, а также фенилаланина (меглитиниды), в своей структуре содержащие т. н. бензамидную группировку (рис. 3). Последняя связывается с SUR c более низкой константой сродства, вот почему новая группа сахароснижающих препаратов, т. н. секретогенов, стимулирует секрецию инсулина значительно слабее и эффект их действия заканчивается быстрее. С другой стороны, с учетом особенностей химической структуры ГБ, содержащего и сульфонилмочевинную, и бензамидную группировки, становится ясно, почему до сего времени он остается “золотым стандартом”, с которым сравнивают активность любого нового сахароснижающего препарата: соединяясь с двумя связывающими
местами на SUR1, ГБ наиболее быстро и мощно способствует закрытию КАТФ-канала, стимулируя деполяризацию мембраны β-клетки, повышение внутриклеточного Са++ и секрецию инсулина.
Риунок 3. Химическая структура ПСМ и секретогенов.
Роль КАТФ-каналов в сердце: экспериментальные данные
КАТФ-каналы присутствуют на плазматических мембранах многих тканей, таких как гладкая и скелетная мускулатура, мозговые и миокардиальные клетки. Естественно предположить возможность негативного влияния вызываемого ПСМ закрытия КАТФ-каналов и повышения концентрации внутриклеточного кальция на состояние миокарда, особенно в условиях ишемии.
Многие авторы изучали роль КАТФ-каналов в ходе адаптации сердечной мышцы к ишемии [28, 29]. При высокой концентрации АТФ в сердечных миоцитах КАТФ-каналы закрыты. Они открываются в различных ситуациях: уменьшение концентрации АТФ, накопление лактата, активация рецептора аденозина A1. В условиях гипоксии или ишемии миокарда снижение внутриклеточного содержания АТФ приводит к открытию КАТФ-каналов. Выход ионов К+ из клеток миокарда инициирует реполяризацию клеточной мембраны, предотвращая вхождение ионов Са++, укорачивая потенциал действия и амплитуду сокращений миокардиоцита, что эффективно снижает потребность миокарда в кислороде и риск последующего его повреждения. Открытие КАТФ-каналов индуцирует феномен т. н. ишемического прекондиционирования (ИПК), которое представляет собой механизм эндогенной защиты миокарда от ишемического повреждения. Впервые явление было описано более 20 лет назад,
когда было обнаружено, что воздействие краткого эпизода ишемии на миокард значительно уменьшало влияние последующей длительной ишемии [30], приводя к значительному снижению размеров формирующегося острого ИМ.
когда было обнаружено, что воздействие краткого эпизода ишемии на миокард значительно уменьшало влияние последующей длительной ишемии [30], приводя к значительному снижению размеров формирующегося острого ИМ.
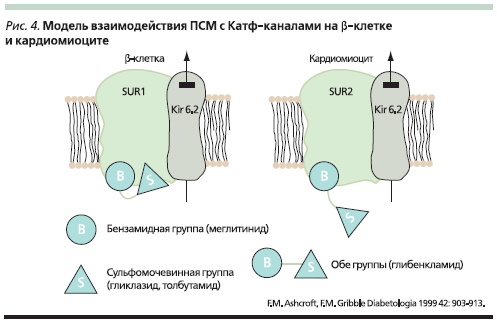
Центральная роль КАТФ-каналов в феномене ИПК подтверждена исследованиями на линии мышей с отсутствием гена Kir6.2, у которых невозможно было индуцировать ИПК [31]. Кроме того, открытие К+-каналов снижает сосудистое сопротивление, повышает коронарный кровоток и снабжение
миокарда кислородом [32]. Таким образом, КАТФ-каналы играют протективную роль в отношении повреждения миокарда при ишемии и вероятность негативного влияния ПСМ на состояние миокарда достаточно велика. Последние достижения в изучении структуры КАТФ-каналов свидетельствуют, что и SUR1- и SUR2-субъединицы канала имеют бензамид-связывающие места, но только SUR1, входящий в структуру КАТФ-канала β-клетки, способен связывать группировку СМ (рис. 4). Из этого логично следует, что препараты, имеющие в своей структуре помимо СМ бензамидную группировку, будут связываться с SUR КАТФ-канала не только на β-клетке, но и на кардиомиоците, а также на гладкомышечной клетке сосудистой стенки; и напротив, препараты, лишенные бензамидной группировки, не способны связываться с SUR2A КАТФ-канала, локализованного в этих тканях (рис. 4).
миокарда кислородом [32]. Таким образом, КАТФ-каналы играют протективную роль в отношении повреждения миокарда при ишемии и вероятность негативного влияния ПСМ на состояние миокарда достаточно велика. Последние достижения в изучении структуры КАТФ-каналов свидетельствуют, что и SUR1- и SUR2-субъединицы канала имеют бензамид-связывающие места, но только SUR1, входящий в структуру КАТФ-канала β-клетки, способен связывать группировку СМ (рис. 4). Из этого логично следует, что препараты, имеющие в своей структуре помимо СМ бензамидную группировку, будут связываться с SUR КАТФ-канала не только на β-клетке, но и на кардиомиоците, а также на гладкомышечной клетке сосудистой стенки; и напротив, препараты, лишенные бензамидной группировки, не способны связываться с SUR2A КАТФ-канала, локализованного в этих тканях (рис. 4).
Таким образом, исследователи вновь вернулись к тому, что ПСМ, в частности ГБ, содержащие бензамидную группировку, могут обладать кардиотоксическим эффектом, способствуя сокращению миокарда за счет стимулированного ими вхождения Са++ внутрь кардиомиоцита и вызывая тем самым ишемию, хотя сродство препарата к SUR2A в 10 раз ниже, чем к SUR1 [33], а продолжительность его связывания с рецептором в сердце значительно короче.
С начала 1990-х гг. опубликовано более 200 исследований, посвященных изучению влияния ГБ – наиболее широко применяющегося представителя II поколения ПСМ – на сердце у животных. Прицельно изучались эффекты ГБ на сосудистый тонус, миокард, его антиаритмическое действие.
Влияние ГБ на миокард в условиях ишемии
Были проведены многочисленные исследования in vitro с использованием изолированного сердца или на животных с открытым сердцем, в которых рассматривался вопрос о способности ГБ увеличивать степень поражения миокардиоцитов при ИМ путем нейтрализации кардиопротективных механизмов, вступающих в действие во время ишемии.
По данным одних авторов, у животных, подвергшихся острой ишемии сердца с последующей реперфузией, введение ГБ увеличивало размер ИМ и сократительную дисфункцию миокарда [34–41]. Другим авторам не удалось обнаружить влияние ГБ на размер ИМ и функциональное восстановление миокарда после фазы ишемии и реперфузии [42, 38, 43, 44–48]. Эти расхождения объясняются методологическими различиями. Наибольшее увеличение размера ИМ отмечено при использовании высоких доз ГБ (3 мг/кг), тогда как низкие дозы (0,3 мг/кг), идентичные терапевтическим при применении в клинике, не оказывали негативного действия [38].
Отмечены и другие противоречия. Так, некоторые авторы сообщили об величении размера ИМ при применении ГБ в дозе 0,15 мг/кг [37], тогда как другими никакого эффекта при его высоких концентрациях (100 мкмоль/л) не отмечено [49]. Потеря способности к ИПК является неизбежной на изолированном сердце. Поэтому возможно, что негативное влияние ГБ может быть связано не столько с подавлением ИПК, сколько с прямым токсическим действием препарата.
Влияние ГБ на ИПК
Многочисленные исследования на животных показали, что ГБ блокирует ИПК через закрытие KATФ-канала в миокарде. У кроликов с ИПК, индуцированном предварительной ишемией в течение 5 минут после 10-минутной реперфузии, зона ИМ уменьшилась на 63 % по сравнению с контрольной группой. У кроликов, получавших ГБ, зона ИМ по размеру была аналогичной таковой в контрольной группе. Таким образом, сокращение в размере зоны ИМ, обусловленное ИПК, нивелировалось у кроликов, получавших ГБ [38]. Этот эффект был подтвержден и на других видах животных: у собак [50], поросят [45]. Вместе с тем у крыс не всегда наблюдали торможение ИПК и блокирование KATФ-канала на фоне ГБ [46, 51]. У кроликов тормозящее действие ГБ на ИПК зависит от используемого анестетика: оно отмечено при применении кетамина и ксилазина, но отсутствовало при использовании пентобарбитала [52]. Эти данные подчеркивают сложность механизмов ИПК. Их экстраполяция на ситуации, связанные с хроническим применением ПСМ, отнюдь не очевидна. Так же не очевидно, что ПСМ влияют на ИПК в ситуации развивающегося ИМ. Только в одном исследовании была проведена оценка сердечного ответа при острой ишемии во время длительного лечения ПСМ крыс со стрептозотоциновым диабетом. Базовые и постишемические функции миокарда у них, как это ни парадоксально, улучшились [53].
Влияние ГБ на аритмию при ишемии миокарда
Желудочковая аритмия – основная причина смерти в острой фазе ИМ. Механизмы этого явления связаны с выходом калия от внутриклеточной среды через открытые KATФ-каналы, что ведет к сокращению длительности потенциала действия, делая сердце более чувствительным аритмогенным
стимулам. ИПК, защищающее сердце от ишемии, может, очевидно, увеличивать риск аритмии. Многими авторами показано, что некоторые вещества, которые открывают KATФ-каналы, увеличивают частоту желудочковых аритмий [54–57], в то время как другие, напротив, снижают ее [58]. Это свидетельствует о комплексном патогенезе аритмии во время миокардиальной ишемии с вовлечением различных механизмов.
стимулам. ИПК, защищающее сердце от ишемии, может, очевидно, увеличивать риск аритмии. Многими авторами показано, что некоторые вещества, которые открывают KATФ-каналы, увеличивают частоту желудочковых аритмий [54–57], в то время как другие, напротив, снижают ее [58]. Это свидетельствует о комплексном патогенезе аритмии во время миокардиальной ишемии с вовлечением различных механизмов.
Антиаритмические свойства ПСМ, включая ГБ, в ситуации острой ишемии миокарда оценивались во многих экспериментальных моделях [39, 57–60]. Большинство этих исследований показало уменьшение частоты случаев желудочковой тахикардии и фибрилляции желудочков. Известно, что в дополнение к благоприятным эффектам на размер ИМ и функцию миокарда ИПК уменьшает риск серьезных желудочковых аритмий во время ишемии и реперфузии [61]. ГБ не блокирует антиаритмический эффект ИПК, как этого можно было бы ожидать. Очевидно, что механизмы, посредством которых снижается выраженность аритмий и уменьшается размер ИМ, различны по своей природе.
В целом в экспериментальных исследованиях in vitro и in vivo влияние ГБ на сердце может быть одновременно и повреждающим, и защитным. При ишемии миокарда ГБ может увеличить зону ИМ, сосудистое сопротивление и блокировать защитный эффект ИПК. Вместе с тем в ситуации острой ишемии препарат обладает антиаритмическими свойствами, что было показано в нескольких исследованиях на животных с СД.
Данные, касающиеся влияния других ПСМ на сердечно-сосудистую систему животных, очень скудны. По сравнению с ГБ гликлазид практически не связывается с SUR2A и SUR2B в культуре Xenope ovocytes [62], однако демонстрирует проаритмогенный эффект у крыс [63], а также уменьшает коронарный кровоток и увеличивает сосудистое сопротивление у собак после внутрикоронарного введения [64]. Следовательно, гликлазид, по-видимому, не влияет на феномен ИПК, но может оказывать проаритмогенный эффект у животных.
Эффекты ПСМ у больных с ишемией миокарда
В перекрестном исследовании, включавшем 19 больных СД2, показано ухудшение функции левого желудочков (по данным Эхо-КГ) при лечении ГБ по сравнению с инсулином [65]. В другой работе при эхокардиографическом обследовании 99 пациентов, получавших ГБ в течение года, исследователи не отметили существенного влияния препарата на массу, фракцию выброса и диастолический объем левого желудочка [66]. При проведении ангиографического исследования показано, что ГБ подавлял защитный эффект ИПК, если он имел место, но не вызывал ухудшения в отсутствие ИПК [67]. В этом исследовании ИПК у пациентов в возрасте старше 65 лет не отмечено.
ГБ не изменяет работу сердца при проведении нагрузочных ЭКГ-тестов [68]. В таких исследованиях ИПК можно вызвать с помощью двух раздельных нагрузочных тестов с интервалом в 15 минут. В разных исследованиях эффекты ГБ различались: в некоторых работах препарат ингибировал ИПК [32, 69, 70], в других – не оказывал подобного действия [71, 72]. Отметим, что только в двух исследованиях участвовали пациенты с СД2, тогда как в остальных работах влияние ГБ на ИПК оценивалось при остром введении препарата у пациентов, не страдающих диабетом. Толерантность к физической нагрузке у больных СД, получавших ГБ, по сравнению с другими ПСМ нарушена не была [70].
Как указывалось выше, одной из основных причин гибели больных в остром периоде ИМ является развитие желудочковой аритмии и фибрилляции за счет снижения концентрации внутриклеточного К+ при ишемии. В этом отношении ГБ обладает наиболее выраженным антиаритмическим эффектом по сравнению с другими ПСМ, включая гликлазид как наиболее аритмогенный препарат и глимепирид, который практически не связывается в миокарде [73, 74]. Антиаритмический эффект ГБ обусловлен его способностью закрывать KATФ-каналы и предотвращать чрезмерную потерю К+ клетками миокарда при выраженной ишемии.
По данным Lomuscio A. и соавт. [89], оценивавших риск развития фибрилляции желудочков у 232 больных СД2 в острой стадии ИМ, наименьшая их частота (1,9 %) отмечена в группе пациентов, получавших ГБ до ИМ, тогда как в группе больных, получавших другую терапию, она составила 7,9 %. Подтверждением этого являются результаты ретроспективного анализа летальности при остром ИМ, проведенного в Австралии на основании оценки 56 715 историй болезни [75]. Уровень летальности составил 12 % у пациентов без СД и 28,1 % больных диабетом (р < 0,001). Частота развития фибрилляции желу-дочков, послужившей причиной смерти у больных СД2, получавших ГБ, была аналогичной таковой у пациентов без диабета (11,8 и 11,0 % соответственно) и меньше, чем у пациентов, принимавших гликлазид (18 %; p < 0,05), а также инсулин (22,8 %; p < 0,05).
Таким образом, многочисленные экспериментальные данные по влиянию ГБ на сердечно-сосудистую систему чрезвычайно противоречивы, что неудивительно, поскольку используемые в эксперименте дозы, как правило, значительно превышают концентрацию препарата в плазме пациентов с СД2, постоянно получающих его. Кроме того, нельзя исключать и того, что гипергликемия сама по себе может предотвращать феномен ИПК [76, 77], а ГБ, как уже упоминалось, не влияет на сердечный ответ в отсутствие ИПК [67]. Поэтому экстраполяция данных о негативном влиянии ГБ на сердечно-сосудистую систему, полученных в эксперименте, а также при обследовании лиц без диабета при остром введении ГБ на пациентов с СД, длительно получающих препарат, является необоснованным.
ПСМ и клинические исходы
Проведенные на сегодня клинические исследования по оценке сердечно-сосудистых исходов у пациентов с СД2 по-прежнему не дают четкого ответа на вопрос: безопасно ли применение ПСМ в этой когорте, с учетом того факта, что сердечно-сосудистая летальность при СД2 в 3 раза превышает таковую в общей популяции? Данные немногочисленных нерандиомизированных ретроспективных исследований чрезвычайно противоречивы: в шведском исследовании, включившем 910 пациентов, наблюдавшихся более 6 лет после ИМ, летальность на фоне ПСМ была значительно ниже, чем при применении комбинации ПСМ с метформином [78], тогда как в канадском исследовании среди 8866 больных, наблюдавшихся после ИМ более 5 лет, напротив, достоверно выше [79]. В популяционном датском исследовании при изучении влияния применения ПСМ на выжимаемость после ИМ достоверных различий между разными препаратами этой группы у 3930 пациентов с СД2
выявлено не было [80].
выявлено не было [80].
Предшествующее развитию ИМ лечение ПСМ не ассоциировалось с увеличением острой (внутрибольничной) летальности после ИМ [81]. Последняя была даже ниже на фоне ПСМ по сравнению с другими методами лечения (10,2 против 16,9 %; p = 0,035) в исследовании, включившем 487 пациентов с СД2 [82]. В другом исследовании с участием 245 пациентов с СД2, госпитали-зированных с острым ИМ, показано, что на фоне предшествующего приема ГБ размер миокардиального некроза не увеличивался [83]. Кроме того, исследование, включившее 245 больных СД2, подвергшихся тромболизису в острой стадии ИМ, отрицательного влияния ПСМ на летальность по сравнению с другими методами лечения не выявило [84].
Многофакторный анализ этих данных показал, что только предшествующее ИМ применение инсулина ассоциировалось со значительно худшим прогнозом выживаемости в остром периоде ИМ и в течение первого года после него. При этом следует учитывать, что применение инсулина необходимо рассматривать как маркер более длительного течения СД2, а большая продолжительность диабета связана с увеличением летальности. Точно так же лечение ПСМ не влияло на тяжесть и прогноз острого ишемического инсульта у 146 госпитализированных пациентов с СД2 [85].
На сегодняшний день проведено только два проспективных рандомизированных исследования с целью определения взаимосвязи между применением ПСМ и сердечно-сосудистой летальностью – UGDP study [17] и UKPDS [6]. Как уже упоминалось, в первом из них выявлено повышение летальности на фоне применения ПСМ толбутамида, сродство которого к SUR2A рецептору KАТФ-каналов оказалось минимальным. В отличие от UGDP в исследовании UKPDS не было выявлено никаких различий в частоте развития ИМ и общей летальности в группах “интенсифицированной терапии”. В конце 10-летнего периода наблюдения перенесенный ИМ имел место только у 90 из 615 пациентов, получавших ГБ (14,6 %), и 100 из 619 (16,2 %), лечившихся хлорпропамидом, т. е. ИМ развился у 190 из 1234 пациентов (15,4 %), принимавших ПСМ. Среди 911 больных, получавших инсулинотерапию, ИМ развился у 149 (16,4 %; р = 0,66). Иными словами, никаких негативных тенденций в отношении ПСМ отмечено не было, хотя, по данным исследования DIGAMI-1, инсулинотерапия на треть сокращала число случаев смерти от ИМ у больных СД2 по сравнению с пациентами, получавшими ПСМ [86]. Следует, однако, отметить, что недавно опубликованные результаты исследования DIGAMI-2 не подтвердили протективного эффекта инсулинотерапии в отношении летальности в ранние сроки и в течение трех лет наблюдения после ИМ. Более того, риск повторного ИМ или инсульта увеличивался на фоне инсулина и не менялся на фоне ПСМ [87]. Сходные результаты были получены в популяционном исследовании, проведенном в Миннесоте (США), при наблюдении за 2189 пациентами, перенесшими ИМ, 386 из которых страдали СД2 и получали либо ПСМ II поколения, либо инсулинотерапию [88].
Недавно завершенные исследования ADOPT, ACCORD, ADVANCE и VADT не ставили своей целью выявить влияние вида получаемого лечения на сердечно-сосудистые исходы, а оценивали значение для гликемического контроля. Тем не менее при анализе полученных результатов не только не было отмечено негативного воздействия ПСМ на кардио-васкулярный прогноз, но, напротив, в исследовании ADOPT показано достоверное снижение кардиоваскулярного риска на фоне применения ГБ в сравнении с росиглитазоном и метформином.
Заключение
Некоторые ПСМ в ряде экспериментальных исследований на животных и у людей оказывали фармакологическое действие на сердце, влияя на феномен ИПК. В настоящее время нет никаких доказательств, что при хроническом применении ПСМ эти эффекты имеют какие-либо клинические последствия. В частности, в исследовании UKPDS не продемонстрировано неблагоприятных сердечно-сосудистых эффектов ГБ или хлорпропамида в течение 11 лет рандомизированного наблюдения. Вместе с тем исследования на людях предполагают, что феномен ИПК ослаблен или отсутствует у пациентов с СД или сердечной недостаточностью [77].
В отсутствие увеличения риска сердечно-сосудистых осложнений, связанного с применением ПСМ, продемонстрированного в специально разработанном и проведенном клиническом исследовании, можно заключить, что феномен ИПК с точки зрения применения препаратов этой группы имеет ограниченное клиническое значение. Также нет никаких свидетельств, будто какой-либо отдельный ПСМ превосходить любой другой при длительном применении в отношении сердечно-сосудистых эффектов. В рамках этой группы лекарств ГБ – наиболее широко используемый во всем мире ПСМ в силу его мощного сахароснижающего эффекта. По-видимому, обоснованно отменять ГБ пациентам с острым коронарным синдромом, а также до и после коронарной ангиографии. Однако в настоящее время не вызывает сомнений, что глибенкламид является эффективным сахароснижающим препаратом, выдержавшим десятилетия успешного клинического применения и продемонстрировавшим положительное влияние на сердечно-сосудистый прогноз.

{kind=link}