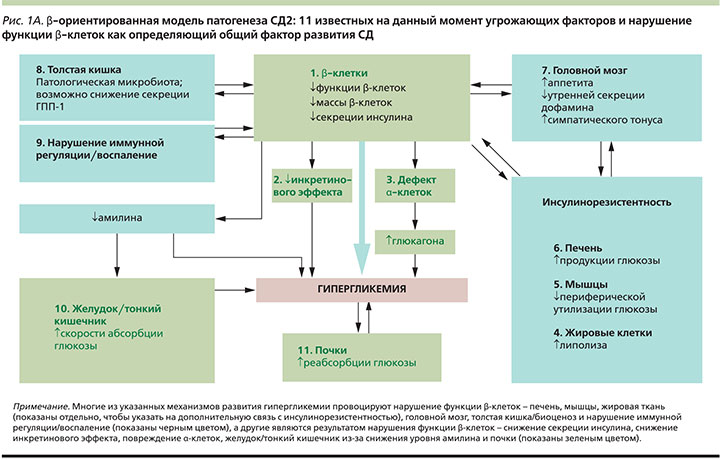
На протяжении многих десятилетий XX в. основной причиной развития сахарного диабета 2 типа (СД2) считался т.н. правящий триумвират, представленный взаимодействием между инсулинорезистентностью периферических тканей, нарушением секреции инсулина β-клетками и повышением продукции глюкозы печенью [1]. Благодаря научным данным, полученным на рубеже веков, произошли очень серьезные изменения в нашем понимании вклада различных факторов регуляции углеводного обмена и терапевтических возможностей влияния на эти факторы. Так, в 2009 г. американским профессором R. De Fronzo впервые была предложена модель, включившая уже «угрожающий октет» ключевых патогенетических звеньев, приводящих к гипергликемии [2]. Стало очевидным, что помимо инсулинорезистентности клеток печени, тканей-мишеней и дисфункции β-клеток важную роль в патогенезе СД2 играют нарушения инкретинового эффекта, гиперпродукция глюкагона а-клетками поджелудочной железы, активация липолиза адипоцитами, усиление реабсорбции глюкозы почками, а также дисфункция нейротрансмиттерной передачи на уровне центральной нервной системы – ЦНС [2]. Данная схема, впервые наглядно продемонстрировавшая гетерогенность развития заболевания, до недавнего времени наиболее четко отражала современные взгляды на патофизиологию СД2. Однако в 2016 г. командой ученых во главе с S.S. Schwartz была предложена в некотором роде «революционная» модель, дополненная еще тремя звеньями развития гипергликемии: системное воспаление, патологическое изменение микрофлоры кишечника и нарушение выработки амилина. Таким образом, на сегодняшний день известно уже о 11 взаимосвязанных механизмах, провоцирующих прогрессирование диабета [3].
Но, пожалуй, самым важным отличием данной модели от всех ранее существовавших является признание именно нарушения функции β-клеток поджелудочной железы фундаментальным определяющим фактором дисрегуляции гомеостаза глюкозы [3]. Как указывают авторы, только β-ориентированная модель предлагает наиболее логичное объяснение взаимосвязи всех 11 известных патофизиологических дефектов СД, т.к. нарушение функции β-клетки находится у истоков манифестации СД2 и коррелирует по принципу обратной связи со всеми остальными механизмами его прогрессирования. И действительно, если обратиться к «натуральному» течению заболевания, клинически выраженный СД возникает в момент, когда комбинированный триггерный механизм генетика/окружающая среда достигает критической точки, в которой происходит достаточно сильное нарушение функции β-клеток [4]. Изначально к факторам развития гипергликемии, которые провоцируют дисфункцию β-клеток, относят органы, непосредственно связанные с инсулинорезистентностью (печень, мышечная и жировая ткани), а также нарушение иммунной регуляции [5–7], патологические изменения микрофлоры кишечника [8–11] и метаболические изменения, регулируемые головным мозгом [12–14]. До тех пор, пока компенсаторные возможности β-клеток позволяют поддерживать секрецию инсулина на достаточном для преодоления инсулинорезистентности уровне, нормальная толерантность к глюкозе сохранена [2, 4, 15, 16]. Однако со временем чувствительность инсулярного аппарата к концентрации гликемии и секреторная способность снижаются, что в результате приводит к развитию гипергликемии. Потребность в инсулине дополнительно повышается также и в результате снижения чувствительности α-клеток, что ведет к неадекватному повышению уровня глюкагона. Эти процессы происходят на фоне нарушения инкретинового эффекта и снижения уровня амилина. Гипергликемия, развивающаяся в результате нарушения функции β-клеток, может вызывать повышение экспрессии белка НГЛТ-2 в почках и приводить к еще большей концентрации глюкозы в крови [17]. Независимо от первопричины гипергликемия приводит к развитию глюкозотоксичности, которая еще больше усугубляет нарушения эндокринной функции поджелудочной железы. Так, нарастающая дисфункция β-клеток определяет скорость прогрессирования заболевания [4]. Следует учитывать, что у разных пациентов наиболее активные механизмы развития гипергликемии могут различаться и, как правило, таких механизмов всегда несколько (рис. 1А).
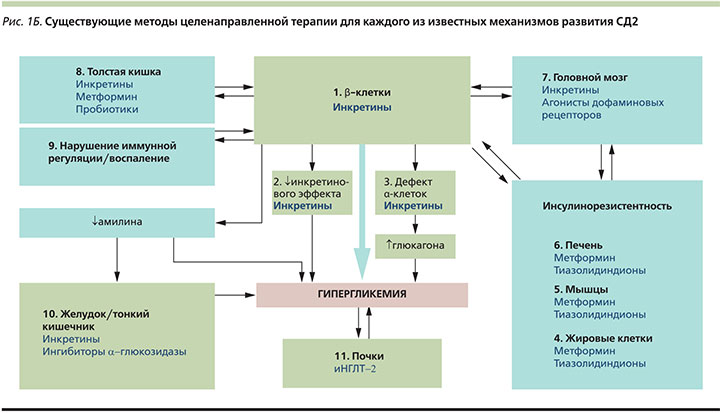
Глубокое понимание многогранности и сложности всех патофизиологических процессов, лежащих в основе СД2, имеет абсолютно практическую ценность и необходимо в первую очередь для успешного управления заболеванием. Учитывая имеющиеся на сегодняшний день знания, β-ориентированная модель может стать концептуальной основой для выбора в пользу терапевтических схем, оказывающих влияние сразу на несколько медиаторов гипергликемии, действуя синергично с целью снижения факторов риска развития сердечно-сосудистых и других заболеваний, а также максимально сохраняя функциональную активность β-клеток. Оптимальная стратегия состоит в применении минимально возможного числа препаратов, при этом воздействующих на максимальное число патогенетических звеньев СД2 [3]. На рис. 1Б показаны механизмы развития гипергликемии и терапевтические возможности современных сахароснижающих средств, также они отражают логику выбора дополнительных механизмов действия в рамках комбинированной терапии.
Как видно на рис. 1Б, применение инкретин-направленной терапии позволяет воздействовать на большинство известных ключевых дефектов, оказывающих влияние на углеводный обмен. При этом в рамках описанной терапевтической концепции особый интерес представляют представители класса ингибиторов дипептидилпептидазы 4-го типа (иДПП-4) ввиду удобства применения таблетированных форм и наличия фиксированных комбинаций с метформином. Ингибирование ДПП-4 направлено на предотвращение инактивации инкретиновых гормонов и восстановление их физиологичных концентраций, что приводит к улучшению нарушенной при СД2 чувствительности β- и α-клеток островков Лангерганса к глюкозе, что ведет к нормализации секреции инсулина и глюкагона как при гипергликемии, так и при гипогликемии [18]. До разработки иДПП-4 возможности корригировать множественные нарушения функции островкового аппарата поджелудочной железы пероральными сахароснижащими препаратами не было. Наряду с этим препараты данной группы обладают и многочисленными вторичными фармакологическими свойствами, позволяя напрямую или опосредованно воздействовать на основные звенья прогрессирования гипергликемии [18]. Столь уникальный механизм действия открывает перед клиницистами все новые перспективы патогенетически обоснованной терапии СД2.
Инкретиновый эффект
Как известно, основное действие глюкозозависимого инсулинотропного полипептида (ГИП) и глюкагоноподобного пептида-1 (ГПП-1), секретируемых L- и К-клетками кишечника, направлено на регуляцию уровня гликемии в ответ на пероральный прием глюкозы [18]. Дефект «инкретинового эффекта» наблюдается уже на стадии нарушения толерантности к глюкозе и особенно выражен у пациентов с ожирением, а одной из основных причин его нарушения служит прогрессирующее снижение массы β-клеток. Несмотря на то что оба гормона стимулируют глюкозозависимую секрецию инсулина, именно ГИП является модулятором активности инкретинов у здоровых людей c нормальной толерантностью к глюкозе. Дефект ГИП-рецепторной передачи и отсутствие его полного эффекта могут играть важную роль на ранних этапах нарушения углеводного обмена и служить объяснением сниженного «инкретинового эффекта» у пациентов с СД2 [19]. При этом глюкагонотропный эффект ГИП остается неизменным [19]. В то же время существуют данные о возможной обратимости инсулинотропного эффекта ГИП у пациентов с СД. Например, в ходе недавних исследований количественной оценки вклада ГПП-1 в регулирование инсулинотропного эффекта у пациентов с СД2 на терапии вилдаглиптином продемонстрировано, что не все инсулинотропные эффекты иДПП-4 связаны только с ГПП-1 [20]. Аналогичные данные были показаны также на фоне терапии ситаглиптином [20]. В связи с этим исследователи полагают, что дополнительный положительный вклад при терапии СД2 иДПП-4 вносит инсулинотропный эффект ГИП в дополнение к уже известному вкладу в регуляцию активности глюкагона при гипогликемии [20].
β-клетки
Еще в ходе доклинических оценок было обнаружено, что инкретины замедляют апоптоз и стимулируют пролиферацию β-клеток, а результаты многочисленных клинических исследований демонстрируют способность иДПП-4 улучшать их секреторную активность и влиять на сохранность функции [3, 18]. Наиболее точную оценку функции β-клеток можно получить, рассчитав скорость секреции инсулина (ССИ) и отношение ССИ/концентрация глюкозы. Согласно имеющимся данным, у пациентов с СД2 этот индекс значимо повышается на фоне приема вилдаглиптина в монотерапии на протяжении 2 лет [21] и при приеме вилдаглиптина в комбинации с метформином [22]. Также при добавлении вилдаглиптина пациентам, не достигшим удовлетворительного контроля гликемии на монотерапии метформином, отмечалось повышение секреции инсулина более чем на 30% уже через 12 недель терапии вилдаглиптином, и этот результат сохранялся на протяжении года исследования [23]. Еще в одном исследовании улучшение функции β-клеток на фоне терапии вилдаглиптином пациентов с недавно диагностированным СД2 сохранялось более 2 лет наблюдения в отличие от группы сравнения, получавшей плацебо [21]. Связь между секрецией инсулина и концентрацией глюкозы включает два компонента – чувствительность и секреторную способность. Чувствительность секреции инсулина к уровню глюкозы особенно важна, поскольку является ранним признаком дисфункции β-клеток при СД2 [18]. Ингибирование ДПП-4 стимулирует секрецию инсулина глюкозозависимым образом именно посредством повышения чувствительности β-клеток к глюкозе. Приведенные данные также указывают на возможность сохранения массы функционирующих на момент постановки диагноза β-клеток, что позволяет приостановить прогрессирующее снижение секреторной способности.
Альфа-клетки
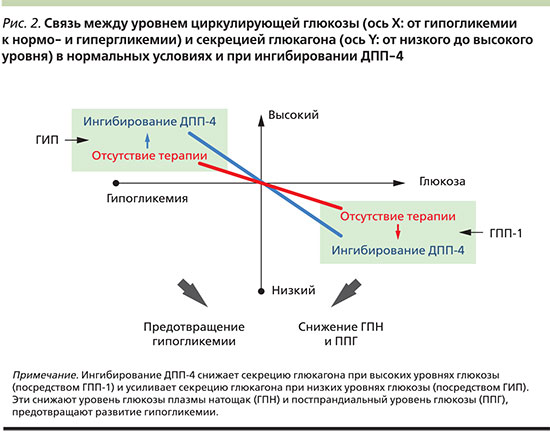
Нормализация секреции глюкагона является важной составляющей терапии СД2, т.к. гиперглюкагонемия способствует прогрессированию гипергликемии натощак и после еды. Влияние иДПП-4 на выработку глюкагона впервые было продемонстрировано в 2004 г., когда выяснилось, что вилдаглиптин подавляет выработку глюкагона в ответ на прием смешанной пищи [24]. Длительное исследование применения вилдаглиптина в комбинации с метформином свидетельствует, что постпрандиальный уровень глюкагона продолжал снижаться и после 2 лет терапии [25], указывая на сохранение глюкагоностатического эффекта с течением времени. Данный эффект, вероятнее всего, объясняется действием ГПП-1, который, согласно имеющимся данным, при СД2 подавляет секрецию глюкагона [26]. В отличие от супрессивного влияния иДПП-4 на неадекватную постпрандиальную секрецию глюкагона препараты данного класса не подавляют контррегуляцию уровня глюкагона в ответ на гипогликемию. Напротив, в ходе исследований было обнаружено, что терапия вилдаглиптином улучшает чувствительность α-клеток к стимулирующему эффекту гипогликемии [27]. Это подтверждает, что ингибирование ДПП-4 влияет на секрецию глюкагона двойственно: когда уровень глюкозы повышается, уровень глюкагона снижается, в то же время при гипогликемии уровень глюкагона увеличивается [27]. Потенциально существенный вклад в контррегуляцию глюкагонового ответа на гипогликемию вносит ГИП, стимулирующий высвобождение глюкагона при нормо- [28] или гипогликемии [29], но не влияющий на секрецию глюкагона при повышенных уровнях глюкозы. Следовательно, влияние ГИП на секрецию глюкагона противоположно влиянию ГПП-1. Таким образом, хотя снижение уровня глюкагона при гипергликемии, по-видимому, опосредуется ГПП-1, вызванная ингибированием ДПП-4 стимуляция секреции глюкагона при гипогликемии может опосредоваться ГИП [18]. В совокупности эти данные указывают на важное двойственное влияние ингибирования ДПП-4 на секрецию глюкагона, опосредуемое как ГПП-1, так и ГИП (рис. 2).
Нарушение иммунной регуляции/воспаление
Было доказано, что наблюдаемое при СД системное воспаление сопровождает стресс эндоплазматического ретикулума на фоне повышенной метаболической потребности в инсулине [30–32]. Таким образом, с клинической точки зрения воспаление оценивается как терапевтическая мишень. Данные клинических исследований демонстрируют, что терапия иДПП-4 оказывает значительное влияние на уровень провоспалительных цитокинов [33–35]. С одной стороны, это свойство обусловлено непосредственным воздействием на рецепторы инкретинов, представленных на поверхности иммунных клеток, с другой – коррелирует с восстановлением физиологической регуляции гликемического профиля на фоне их применения [36–38]. При этом и внутри класса наблюдаются некоторые различия во влиянии на показатели воспаления и оксидативного стресса. Так, терапия вилдаглиптином по сравнению с ситаглиптином обеспечивала более стабильный уровень глюкозы крови в течение суток, что сопровождалось позитивным влиянием на маркеры свободнорадикальных процессов и воспаления [35].
Головной мозг, ЦНС
Гипоталамус принимает важнейшее участие в нейроэндокринной регуляции процессов метаболизма, оказывая влияние и на функцию поджелудочной железы. Сигналы от кишечника поступают в головной мозг через волокна блуждающего нерва. Так, инкретины, синтезируемые в клетках кишечника, через нервную систему способны регулировать функционирование β- и α-клеток [39, 40]. Как известно, при СД2 отмечается дисфункция нейротрансмиттеров [2]. В отличие от аналогов ГПП-1, которые напрямую связываются с ГПП-1-рецепторами в периферических тканях (головной мозг и поджелудочная железа), ингибирование ДПП-4 может приводить к увеличению концентрации ГПП-1 непосредственно в кишечнике, после чего ГПП-1 идентифицируется афферентными нервными волокнами блуждающего нерва в кишечнике и воротной вене. А далее уже интеграция нервной системы координирует метаболические эффекты в периферических тканях через эфферентные волокна блуждающего нерва [41]. То есть при введении ГПП-1 в фармакологической дозе ГПП-1 работает как гормон, а в физиологической концентрации при ингибировании иДПП-4 – как нейротрансмиттер [41]. Данные свойства, вероятно, также вносят свой вклад в нормализацию физиологической регуляции уровня гликемии у пациентов с СД на фоне применения иДПП-4.
Микробиота
Пристальное внимание в диабетологии последнее время уделяется и состоянию микрофлоры кишечника [8–11]. Была продемонстрирована достоверная связь между состоянием микробиоты как с ожирением, так и с СД1 и СД2, что может помочь объяснить тот факт, что СД с выраженными симптомами развивается только у определенной доли людей с избыточной массой тела [9–11].
Дисбаланс микрофлоры приводит к воспалению, секреции цитокинов, инсулинорезистентности и снижению секреции ГПП-1 [42]. В результате патологическая микробиота, вероятно, оказывает отрицательное влияние на функционирование β-клеток и через нарушение передачи сигнала в головной мозг. Применение же иДПП-4 способствует поддержанию физиологических концентраций ГПП-1 и, как уже было указано, может приводить к улучшению передачи инкретинового сигнала по блуждающему нерву [40–41].
Снижение секреции амилина
Снижение выработки амилина, также являясь следствием нарушения функции β-клеток, приводит к ускорению опорожнения желудка и оказывает определенное влияние на повышение абсорбции глюкозы в тонком кишечнике с сопутствующим повышением постпрандиальных уровней глюкозы [3, 43]. При этом многие из свойств амилина схожи со таковыми ГИП и ГПП-1, в связи с чем теоретически инкретины могут использоваться для воздействия на этот механизм развития гипергликемии [44]. Кроме того, терапия иДПП-4, улучшая чувствительность β- и α-клеток и нормализуя постпрандиальную гликемию, опосредованно может нивелировать отрицательное влияние дефекта секреции амилина на дальнейшее прогрессирование СД2.
Инсулинорезистентность
Согласно фундаментальным представлениям о патогенезе развития СД2, резистентность периферических тканей (мышечная, жировая и ткани печени) к действию инсулина служит одним из ключевых звеньев метаболических нарушений, не воздействуя на которое любая терапия обречена на провал. Без сомнения, препаратом выбора для коррекции инсулинорезистентности является метформин. При этом иДПП-4 в свою очередь оказывают влияние на инсулинорезистентность, улучшая функцию β-клеток, снижая уровень провоспалительных цитокинов и окислительного стресса [18, 35, 36], что обеспечивает дополнительные преимущества при добавлении к метформину. Так, результаты исследований демонстрируют достоверно большее снижение инсулинорезистентности (индекс HOMA-IR – Homeostasis Model Assessment of Insulin Resistance), а также улучшение функции β-клеток (HOMA-β) на фоне терапии вилдаглиптин+метформин по сравнению с монотерапией метформином [49]. Важным свойством иДПП-4 является отсутствие прибавки массы тела на фоне их применения с учетом глюкозозависимого действия на β- и α-клетки поджелудочной железы, что обусловливает минимальный риск развития гипогликемии и отсутствие проблемы «защитного» заедания» [18]. Кроме того, известны вторичные фармакологические механизмы действия иДПП-4, включающие внепанкреатическое влияние на метаболизм липидов и утилизацию глюкозы в мышечной ткани [18]. Некоторые данные демонстрируют, что терапия вилдаглиптином усиливает постпрандиальный липолиз в жировой ткани и увеличивает постпрандиальное окисление жиров в мышцах [45]. Более того, на фоне применения иДПП-4 наблюдалось снижение общей концентрации триглицеридов и хиломикронов в сыворотке крови, что отражает снижение уровня аполипопротеина B-48 (Apo B-48) и холестерина в составе хиломикронов [46–48]. Так как хиломикроны являются классом липопротеинов, образующихся в тонком кишечнике в процессе всасывания экзогенных липидов, эти результаты могут свидетельствовать о том, что вилдаглиптин оказывает тормозящее действие на всасывание жира в кишечнике [18].
Таким образом, еще раз взглянув на все возможности инкретин-направленной терапии, можно заключить, что своевременное назначение комбинации препаратов иДПП-4+метформин представляет собой элегантную схему управления СД2, эффективно воздействующую на 10 из 11 известных патогенетических звеньев прогрессирования заболевания. Данная схема отражает взгляды ведущих диабетологов, поддерживающих прогрессивную идею «патофизиологического подхода», когда для достижения успеха терапия должна не просто быть направлена на снижение уровня гликозилированного гемоглобина (HbA1c), но и оказывать влияние на большинство факторов развития диабета и модификацию его естественного течения [4]. Вместе с тем все большей критике подвергается традиционный «ступенчатый» подход к терапии СД2, представляющий собой стратегию «лечение до неудачи». Инициацию и интенсификацию терапии необходимо начинать как можно раньше с целью профилактики либо замедления прогрессирования недостаточности β-клеток [4, 50]. Одним из ключевых аспектов для выбора начальной комбинации сахароснижающих препаратов должна являться физиологичность назначаемой терапии, т.к. смысл раннего интенсивного старта заключается в попытке замедлить темпы заболевания. В этом отношении большой интерес представляет многонациональное многоцентровое пятилетнее исследование VERIFY, стартовавшее в 2014 г., цель которого состоит в оценке клинических преимуществ комбинированной терапии метформином и вилдаглиптином пациентов с недавно диагностированным СД2 и исходным уровнем HbA1c 6,5–7,5%. Это тот уровень, когда, согласно существующим алгоритмам, не требуется назначения комбинированной сахароснижающей терапии [50]. Результаты исследования позволят получить ответ на вопрос, может ли раннее назначение комбинации ингибиторов ДПП-4 с метформином уменьшить скорость прогрессирования СД.
Заключение
Итак, сегодня нам известны уже 11 звеньев патогенеза СД2. Предложенная β-ориентированная модель и в дальнейшем будет дополняться на основе актуальных знаний и новых открытий, но уже сейчас должна стать концептуальной основой в выборе предпочтительных фармакологических средств, действие которых будет направлено на активные механизмы развития гипергликемии у каждого конкретного пациента. Идеальная парадигма патофизиологического подхода должна состоять в коррекции множественных нарушений, лежащих в основе прогрессирования СД2, а разные классы препаратов рассматриваться в качестве взаимодополняющих вариантов в составе ранней комбинированной терапии, а не как дополнительный резерв уже на этапе неизбежной терапевтической неудачи. При этом выбор схем терапии необходимо проводить с учетом их влияния на функцию β-клетки.
