Введение
Крапивница и уртикарный васкулит – кожные заболевания, часто сопровождающиеся зудом и проявляющиеся эритематозными (красными, розовыми) волдырными высыпаниями (при крапивнице они исчезают в течение 24 часов, при уртикарном васкулите сохраняются дольше). Уртикарии относятся к числу наиболее распространенных болезней кожи. Так, в течение жизни 0,5–1,0 % всей популяции страдает хронической крапивницей [1]. Нередко уртикарии являются симптомом другого заболевания, поэтому изучение проблемы актуально не только для дерматологов и аллергологов-иммунологов, но и для терапевтов, а также врачей других специальностей. Наиболее частые причины хронической крапивницы (ХК) представлены в табл. 1.
Несмотря на то что на долю т. н. других причин ХК приходится менее 10 % случаев, следует помнить, что ХК может быть проявлением потенциально опасных заболеваний внутренних органов, в связи с чем пациенты с рецидивирующими уртикарными высыпаниями нуждаются в тщательном обследовании.
Перечень возможных системных заболеваний обширен и включает:
- аутоиммунные заболевания (болезнь Стилла у взрослых, системная красная волчанка);
- гематологические заболевания (макроглобулинемия Ванденстрема, лимфомы, POEMS-синдром);
- наследственные аутовоспалительные заболевания (Синдром Маккла–Уэллса, семейная холодовая крапивница);
- сóлидные новообразования;
- другие заболевания: криоглобулинемия, мастоцитоз, синдром Шницлер и др.
Первое описание синдрома Шницлер (СШ) было представлено в 1972 г. французским дерматологом Лилиан Шницлер [3]. Данный синдром представляет собой сочетание ХК и моноклональной иммуноглобулин М-(IgM)-гаммапатии, рассматривается как вариант приобретенного в зрелом возрасте аутовоспалительного синдрома. Заболевание относится к числу редких. С момента описания данного синдрома было зарегистрировано около 200 случаев [4].
Главные клинические проявления СШ включают уртикарную сыпь, рецидивирующую лихорадку, боли в мышцах, костях и/или суставах, а также увеличение лимфатических узлов. Моноклональная гаммапатия IgM (или реже IgG) является биологическим маркером заболевания. Кроме того, отмечается стойкое повышение уровня маркеров воспаления, таких как лейкоциты, СОЭ, С-реактивный белок (СРБ). Для диагностики синдрома используют критерии, предложенные в 2001 г. Д. Липскером и соавт. [5] (табл. 2).

СШ имеет рецидивирующее течение с периодами спонтанной ремиссии различной длительности. Традиционное лечение включает антигистаминные средства, используемые для купирования кожных высыпаний и зуда, а также противовоспалительные и иммуносупрессивные средства для подавления системных проявлений. Однако последние обычно малоэффективны и обладают серьезными побочными эффектами. В единичных наблюдениях показана эффективность глюкокортикоидов, а также колхицина, циклоспорина А, пефлоксацина [4], ритуксимаба. Наиболее эффективным при СШ считается применение антагониста рецептора интерлейкина-1 – анакинры, однако использование этого препарата в России ограничено.
У 15–20 % больных СШ в течение 10–20 лет наблюдения развиваются различные лимфопролиферативные заболевания [6], а также АА-амилоидоз. Таким образом, возможность малигнизации заболевания и появление амилоидоза обусловливают необходимость тщательного наблюдения таких пациентов на протяжении многих лет.
В связи с редкостью заболевания и отсутствием сведений о СШ в отечественной литературе мы приводим описание двух случаев с различными клиническими особенностями и ответом на терапию.
Клинический случай № 1
Пациент П. 42 лет, по профессии слесарь, житель Башкирии, поступил в Клинику нефрологии, внутренних и профессиональных болезней им. Е.М. Тареева УКБ № 3 в октябре 2007 г. с жалобами на высыпания, локализующиеся по всему кожному покрову, сопровождающиеся незначительным зудом, а также повышением температуры тела до 37,5 °С.
Из анамнеза заболевания: считает себя больным с декабря 2004 г., когда впервые на коже нижних конечностей появились ярко-красные высыпания по типу крапивницы, в последующем высыпания распространились на верхние конечности, а затем – на все тело. Они были склонны к слиянию, не сопровождались зудом, сохранялись в течение 1–2 суток и проходили бесследно (рис. 1 а, б). Кроме этого больного беспокоило повышение температуры тела до 37 °С.
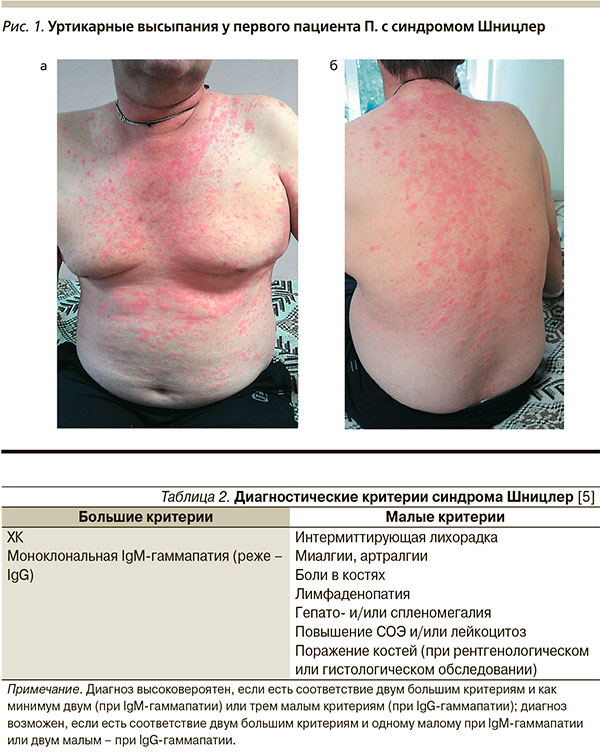
Дерматологом по месту жительства кожные изменения расценивались как крапивница, проводилось лечение антигистаминными препаратами, глюконатом кальция без существенного эффекта. Эпизоды крапивницы рецидивировали каждые 3–4 дня. Пациент продолжал работать. С 2006 г. кожные высыпания стали носить более распространенный характер, сопровождались повышением температуры тела до 38,5 °С, ознобами, появились боли в мышцах ног, коленных суставах. Пациент прекратил работать. При обследовании: СОЭ – 36 мм/ч, лейкоцитоз Ц до 10 × 109∕л, положительная реакция на СРБ. В результате обследования по месту жительства, включившего гастро-, колоноскопию, стернальную пункцию, неоднократные посевы крови, исследования крови на различные инфекционные заболевания, не выявлено связи кожного синдрома с инфекционными, гематологическими, аутоиммунными заболеваниями, опухолями с паранеопластическим синдромом. В течение последующего времени практически постоянно сохранялась высокая воспалительная активность. Проводились короткие курсы терапии преднизолоном в дозе до 30 мг/сут с кратковременным положительным эффектом.
При первичном осмотре кожный патологический процесс носил распространенный характер, были выявлены высыпания по типу крапивницы на руках, ногах, по всему туловищу, склонные к слиянию. Отклонений со стороны дыхательной, сердечно-сосудистой и мочевыделительной систем, желудочно-кишечного тракта не выявлено. Температура тела – 37,5 °С.
Состояние периферической нервной системы без особенностей.
Для уточнения диагноза проведены различные обследования. Биохимические показатели крови, уровни комплемента, криоглобулинов, антинейтрофильных цитоплазматических антител оказались без существенных отклонений от нормы. Маркеры вирусных гепатитов были отрицательными. Сохранялись умеренный лейкоцитоз, повышенная СОЭ, высокий уровень СРБ; обнаружено двукратное повышение уровня IgM. При иммуноэлектрофорезе сыворотки крови выявлен М-градиент в γ2 зоне, образованный парапротеином IgМ κ (7,3 % от общего белка сыворотки или 4,7 г/л), а также следовой М-градиент, образованный парапротеином IgG λ, воспалительная диспротеинемия. Белок Бенс–Джонса не обнаружен. При гистологическом исследовании пораженного участка кожи выявлены гиперкератоз, разрыхление и отек дермально-эпидермальных стыков, экссудативно-пролиферативные васкулиты, небольшие лимфопролиферативные инфильтраты периваскулярно; амилоида не найдено; картина васкулита в дерме. При остеосцинтиграфии с пирофосфатом технеция обнаружены очаги патологической гиперфиксации радионуклидов в нижних конечностях. При трепанобиопсии подвздошной кости признаки специфического поражения костного мозга не выявлены.
Таким образом, в процессе тщательного обследования исключены признаки общеизвестных аутоиммунных, инфекционных, гематологических заболеваний, связанных с ХК. В то же время выявленные высыпания, подобные крапивнице (уртикарный васкулит), моноклональная IgМ-гаммапатия в сочетании с лихорадкой, мышечно-суставным синдромом, стойким повышением СОЭ, лейкоцитозом позволяют диагностировать СШ. Все вышеуказанные признаки соответствуют диагностическим критериям Д. Липскера [4].
В связи с выраженными системными проявлениями была начата терапия преднизолоном по интермиттирующей схеме (чередование приема 20 и 30 мг/сут) в сочетании с гидроксихлорохином (ГХХ, 400 мг/сут). В течение нескольких дней после начала терапии отмечено существенное улучшение самочувствия: нормализовалась температура тела, уменьшилась выраженность высыпаний, купированы боли в ногах. Больной был выписан под амбулаторное наблюдение. В последующем в течение полугода состояние оставалось стабильным, однако при попытках снижения дозы глюкокортикоидов отмечались рецидивы симптомов. Пациент возобновил трудовую деятельность.
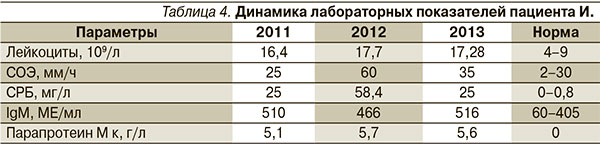
Невозможность снижения дозы преднизолона свидетельствовала о недостаточной эффективности ГХХ и обусловила поиск альтернативных препаратов базисной терапии. Обсуждалась возможность назначения колхицина, циклоспорина А, интерферона α. Была предпринята попытка добавления к терапии сульфасалазина (до 2 г/сут), не давшая положительного результата. В результате литературного поиска обнаружено указание на эффективность применения пефлоксацина [7]. С учетом данного факта, а также выявления в общем анализе мочи пациента лейкоцитурии, наиболее вероятно, в результате бессимптомной инфекции мочевыводящих путей больному был назначен пефлоксацин в дозе 800 мг/сут. После этого было отмечено снижение выраженности высыпаний и улучшение общего самочувствия. Терапия была продолжена и после разрешения лейкоцитурии. Постепенно удалось снизить дозу преднизолона до поддерживающей (10 мг/сут). Переносимость пефлоксацина при длительном приеме была удовлетворительной. Отмечена повышенная фоточувствительность (покраснение кожи лица и открытых участков тела после пребывания на солнце), в связи с чем рекомендовано также продолжение приема ГХХ (доза снижена до 200 мг/сут). Следует отметить, что несмотря на уменьшение выраженности лихорадки и кожных высыпаний, сохранялись стойкие изменения лабораторных показателей: высокая воспалительная активность (повышение СОЭ, высокий уровень С-реактивного белка), при повторном иммуноэлектрофорезе выявлен IgМ κ приблизительно в том же количестве (7,5 % от общего белка сыворотки, или 4,9 г/л). Кроме того, высыпания рецидивировали непрерывно, появилась клиника периферической невропатии. В 2010 г. после повторной трепанобиопсии к терапии добавлен ритуксимаб (2000 мг, затем по 1000 мг каждые 6 месяцев) – моноклональные антитела к CD20-антигену В клеток. В ответ на терапию ритуксимабом отмечено существенное улучшение состояния пациента, снижение воспалительной активности и уровня моноклональной секреции (табл. 3). Переносимость лечения препаратом на протяжении 3 лет удовлетворительная.
Клинический случай № 2
Пациент И. 50 лет, по профессии охранник, житель Москвы, обратился в клинику кожных и венерических болезней им В.А. Рахманова в феврале 2013 г. с жалобами на высыпания на коже туловища, конечностей, периодически возникающий невыраженный зуд кожи; боль в костях голеней, верхних конечностей; слабость, потливость; увеличение шейных, паховых лимфоузлов; периодически возникающую изжогу, ноющие боли в области живота; озноб, повышение температуры до 37,4 °С.
Анамнез заболевания: считает себя больным с конца 2007 г., когда впервые отметил появление на коже туловища и конечностей высыпания по типу крапивницы. По этому поводу наблюдался в поликлинике по месту жительства у дерматологов с диагнозом «крапивница» и «аллергический дерматит». По назначению врачей принимал антигистаминные и антибактериальные препараты, но эффекта от лечения не отмечал. С другой стороны, на фоне строгого соблюдения гипоаллергенной диеты в течение 1–2 недель высыпания исчезали или заметно уменьшались. Другого лечения не получал.
Боли в костях голеней, предплечий, периодическую потливость отмечает с 2009 г. В течение длительного времени (с 2009 по 2012 г.) консультировался у различных специалистов (флеболога, хирурга, гематолога, ревматолога, онколога) с диагнозом «крапивница» и подозрением на системный процесс. В 2012 г. при осмотре онкологом было обнаружено увеличение паховых лимфоузлов до 1,5 см в диаметре, подвижных, не спаянных между собой. По результатам анализа крови выявлено повышение лейкоцитов, СОЭ, СРБ, общего IgM, обнаружен парапротеин (табл. 4). Кроме этого имело место увеличение ревматоидного фактора до 436 ЕД/мл (норма – 0–200).
В октябре 2011 г. пациенту проведена радионуклидная визуализация скелета. Радионуклидная картина оказалась характерной для пролиферативного процесса в костном мозге с ярковыраженной реакцией костной ткани позвонков костей таза, грудины, ключицы, правой лопатки, правой плечевой, бедренных и больших берцовых костей. Неоднократно осуществлялась трепанобиопсия костного мозга. В последнем заключении (ноябрь 2011 г.) указано на отсутствие изменений в биоптате костного мозга, характерных для миелопролиферативного заболевания; имеющиеся изменения соответствовали картине вторичных нарушений гемопоэза.

В течение первой половины 2012 г. больной наблюдался в НИИР РАМН, консультирован аллергологом, гематологом, ревматологом. Специфической патологии не выявлено. Проведена трепанобиопсия костного мозга (от 31.01.12) и радиоизотопное исследование костей скелета (от 13.02.12). Заключение трепанобиопсии: убедительных морфологических признаков миелопролиферативной опухоли не найдено. Заключение радиоизотопного исследования: определено повышенное накопление радиофармпрепарата в костях свода черепа, ключицах, длинных трубчатых костях, проксимальных отделах плечевых костей. Данную картину следует дифференцировать между остеодистрофией и вторичными изменениями в скелете.
В июне 2012 г. выполнена биопсия кожного лоскута, по результатам гистологического исследования материала в РОНЦ им. И.М. Блохина, данных за опухолевый процесс не выявлено.
Согласно заключению иммуногистохимического исследования белков сыворотки и мочи от 23.05.12, выявлена моноклональная секреция парапротеина М κ (5,7 г/л), составляющего 8,2 % от общего белка сыворотки крови, уровень поликлональных иммуноглобулинов не снижен, секреция белка Бенс–Джонса не выявлена, обнаружены признаки воспалительной диспротеинемии.
При проведении мультиспиральной компьютерной томографии (март 2012 г.) отмечено увеличение лимфоузлов средостения, подмышечных областей, брюшной полости, забрюшинного пространства, подвздошных и паховых областей, эмфизема легких, гепатомегалия.
В последующем больной был госпитализирован в отделение онкологии ФГКУ ГКВГ ФСБ России (с 30.07.12 по 25.08.12). При осмотре пациента выявлено увеличение паховых лимфоузлов (до 1,5 см), подвижных, слабоболезненных при пальпации. При осмотре кожи верхних и нижних конечностей, туловища определены множественные папулезные мягкоэластичные образования неправильной формы размером от 1 до 5 см в диаметре, сливного характера, безболезненные при пальпации. В анализе крови от 31.07.12: лейкоциты 18,6 × 109/л (норма – 4,0–9,0 × 109/л), СОЭ – 28 мм/ч (норма – 0–20 мм/ч), фибриноген – 7,85 г/л (норма – 2–4 г/л).
Интересно, что с 02.06.11 по 08.08.12 у пациента неоднократно выявлялся высокий уровень общего IgE, имевший некоторую тенденцию к снижению с течением времени (табл. 5). При этом уровень специфических IgE к различным аллергенам был отрицательным.
На основании клинической картины и результатов исследований был выставлен диагноз «лимфопролиферативное заболевание с поражением регионарных лимфоузлов, кожи», не подтвердившийся впоследствии. Была проведена пункция пахового лимфоузла справа. Заключение иммуногистохимического исследования от 27.08.12: в лимфатическом узле имеется реактивная поликлональная лимфоидная гиперплазия.
В отделении пациенту проведена ферментная, антигистаминная, антибактериальная терапия – без эффекта в отношении кожного процесса. На фоне проведенного консервативного лечения (омепразол, висмута трикалия дицитрат, диета) явления эрозивного антрального гастрита (изжога, ноющие боли в области живота) были купированы. После назначения диеты пациент отметил уменьшение кожных высыпаний.
Результаты очередного иммуногистохимического исследования белков сыворотки и мочи оказались без динамики по сравнению с предыдущим.
С ноября 2012 г. пациент наблюдается у онколога, гематолога ФГБУ «РОНЦ им. Н.Н. Блохина» РАМН, где для определения диагноза заболевания проведены различные исследования. Заключение рентгенографического исследования от 09.11.12: остеохондроз на уровне Th4–Th11, левосторонний сколиоз грудного отдела позвоночника первой степени. В поясничном отделе начальные проявления остеохондроза на уровне L4–L5.
Заключение исследования пунктата костного мозга от 21.11.12: костный мозг представлен примерно равным соотношением жировой и кроветворной тканей. Видны элементы всех ростков гемопоэза. Число мегакариоцитов умеренно увеличено, среди них встречаются микро-, гиполовулярные формы. Умеренно расширен красный росток, гранулоцитарный, скорее всего без особенностей. Имеются многочисленные макрофаги, в них – признаки фагоцитоза. Изменения не соответствуют какой-либо определенной нозологической единице. Миелограмма: отпечаток трепанобиоптата клеточный; гранулоцитарный ряд расширен за счет увеличения числа молодых и зрелых форм нейтрофилов; красный росток сужен; мегакариоциты в достаточном количестве. Заключение цитогенетического исследования клеток костного мозга от 21.11.2012: при стандартном цитогенетическом исследовании патологические изменения не выявлены. Заключение ДНК-диагностики мутации гена JAK2 V617F – маркера хронических миелопролиферативных заболеваний: отрицательно.
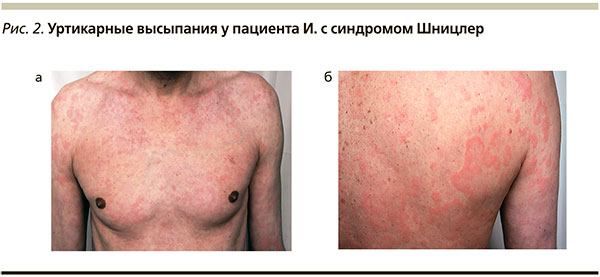
В Клинике кожных и венерических болезней им. В.А. Рахманова (февраль 2013 г.) больной был проконсультирован врачом-дерматологом и аллергологом-иммунологом. При осмотре кожи туловища, конечностей отмечены рассеянные множественные уртикарные элементы различного размера розового цвета (рис. 2 а, б). Присутствовал слабый зуд кожи верхних конечностей. Отеков не было. Наблюдалось незначительное увеличение шейного лимфоузла справа, он был безболезненным, не спаянным с кожей. Других патологических изменений при осмотре выявлено не было. Высыпания сохранялись постоянно, периодов «чистой кожи» не было. Отдельные элементы держались 1–3 суток, исчезая без остаточных явлений. Зуд появлялся спонтанно, был невыраженным, на любых участках кожи, в т. ч. на коже, свободной от высыпаний. Ангиоотеков больной не отмечал. Персональный аллергологический анамнез не отягощен, семейный: у сына – астма, у внука – пищевая аллергия. Лечения по поводу заболевания на момент осмотра не получал, диету не соблюдал. Пациент отмечал обострение крапивницы при применении препарата из группы нестероидных противовоспалительных средств (НПВС). Тест с аутологичной сывороткой оказался отрицательным. Был предположен диагноз «уртикарный васкулит как проявление общего системного заболевания».
Пациент был направлен к гематологу ФГБУ «Гематологический научный центр» Минздрава РФ, где на основании клинической картины и выявленных изменений в результатах электрофореза белков сыворотки (обнаружен парапротеин М κ, который составлял 7,4 % от общего белка сыворотки крови, или 5,6 г/л) был установлен диагноз «синдром Шницлер».
Заключение микроскопического исследования биоптата кожи из мест высыпаний от 03.04.13: в коже картина неопухолевого дерматоза/дерматита (аутоиммунного генеза – ранние проявления васкулита?).
Таким образом, так же как и у пациента из предыдущего наблюдения, в процессе тщательного обследования исключены признаки общеизвестных аутоиммунных, инфекционных, гематологических заболеваний, связанных с ХК или уртикарным васкулитом.
В то же время выявленные ХК (уртикарный васкулит), моноклональная IgМ-гаммапатия в сочетании с болью в костях, лимфаденопатией, гепатомегалией, стойким повышением СОЭ и лейкоцитозом позволяют диагностировать СШ. Все вышеуказанные признаки соответствуют диагностическим критериям Д. Липскера [4].
Обсуждение
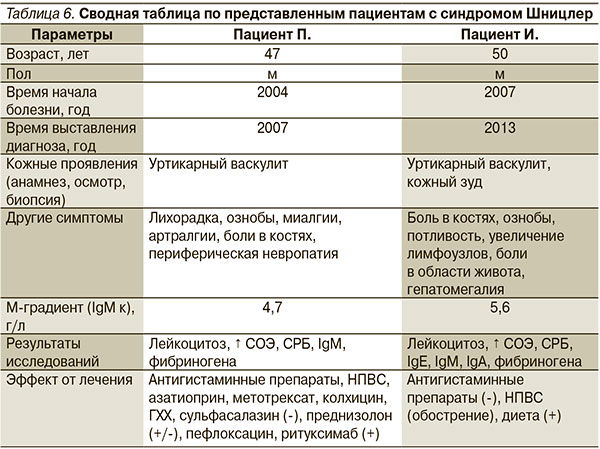
В описанных нами двух случаях СШ есть как сходства, так и различия. Наблюдения объединяют мужской пол, общая возрастная группа, схожие кожные проявления болезни, наличие моноклональной IgM-гаммапатии и повышение уровней маркеров воспаления в крови. Кроме этого и в том, и в другом случае выставление правильного диагноза было отсрочено на несколько лет (на 4 года и 6 лет соответственно). С другой стороны, случаи отличаются внекожными симптомами, а также увеличением уровней IgA и IgE и эффективностью диеты у второго пациента (табл. 6).
Значительное увеличение уровня общего IgE в отсутствие вторичных причин (аллергии, паразитарной инвазии и т. д.) является интересной особенностью второго случая и не было ранее описано в литературе. Из анамнеза видно, что в июне 2011 г. уровень IgE составлял 2156 МЕ/мл (норма – до 100) и постепенно уменьшался в течение нескольких лет.
Дифференциальная диагностика СШ проводится с криопирин-ассоциированными периодическими синдромами, системной красной волчанкой, гипокомплементарным уртикарным и криоглобулинемическим васкулитами, инфекционными болезнями, хронической идиопатической крапивницей и болезнью Стилла для взрослых. Все эти заболевания были исключены у наших пациентов в процессе диагностического поиска.
Считается, что для СШ более характерно появление ХК, чем напоминающего ее уртикарного васкулита.
В наших наблюдениях в обоих случаях уртикарный васкулит был подтвержден результатами гистологического иссле-дования («золотой» стандарт диагностики васкулита) и особенностями кожного процесса (длительное сохранение высыпаний, отсутствие эффекта от антигистаминного лечения). Многие специалисты выделяют уртикарный васкулит как одну из форм крапивницы.
Тем не менее пока не ясно: влияет ли наличие уртикарного васкулита, а не крапивницы на течение основного заболевания и если да, то каким образом?
Единого подхода к лечению заболевания не найдено. В единичных наблюдениях показана эффективность глюкокортикоидов, а также колхицина, циклоспорина А, интерферона α, пефлоксацина и ритуксимаба. Определенные надежды возлагают на биологический препарат — антагонист рецепторов интерлейкина-1 (анакинра). В первом клиническом случае мы отметили эффективность пефлоксацина и ритуксимаба, что привело к хорошему контролю заболевания.
Эффективность пефлоксацина поднимает интересные вопросы о патогенезе СШ. Некоторые авторы придают большое значение фиксации в капиллярах и венулах дермы макромолекулярных комплексов IgМ с последующим развитием неспецифической воспалительной реакции [8]. Другие отмечают, что иммунофлуоресцентные исследования позволяют обнаружить кожные депозиты IgM только у некоторых пациентов [7]. Альтернативная гипотеза основную патогенетическую роль отводит продукции одного или нескольких цитокинов или хемокинов. Данный подход позволяет объяснить клиническую эффективность пефлоксацина, не связанную с изменением уровня IgM. Пефлоксацин, как и другие фторхинолоны, реализует свой бактерицидный эффект посредством ингибирования ДНК-гиразы (топоизомераза II типа), скорее всего за счет связывания с ДНК. Помимо антибактериальных свойств фторхинолоны обладают также способностью влиять на иммунные и воспалительные реакции посредством воздействия на механизмы регуляции образования таких цитокинов, как интерлейкины-1 и -2 и их рецепторы, интерферон γ, гранулоцитарно-макрофагальный колониестимулирующий фактор и интерлейкин-13 [9]. Тот факт, что пефлоксацин является наиболее сильным среди фторхинолонов [7], объясняет отсутствие эффекта при использовании других препаратов этой группы.
Данные литературы по использованию ритуксимаба противоречивы.
В одних исследованиях эффекта от применения препарата не отмечали [10, 11], в других – такой эффект был [12, 13]. Мы продемонстрировали возможность его использования первым пациентом с удовлетворительными клиническими и лабораторными результатами.
Интересно, что первому пациенту назначали диету, а также массу различных препаратов (НПВС, азатиоприн, метотрексат, колхицин, ГХХ), в первую очередь цитостатических, без какого-либо эффекта, а у второго больного соблюдение строгой элиминационной диеты привело к полному очищению кожи от высыпаний, но сохранению системных симптомов в отсутствие ответа на другое лечение.
Важно, что эффективность диеты, так же как повышение уровня общего IgE, не характерны для СШ и ранее не были описаны в литературе.
Таким образом, приведенные клинические случаи указывают на то, что при одном и том же заболевании терапевтическая тактика может сильно различаться. В то время как одному пациенту для контроля кожного процесса может быть достаточно диеты, для лечения другого может потребоваться время, чтобы подобрать эффективный препарат.
Течение СШ достаточно благоприятное: 15-летняя выживаемость превышает 90 %. Однако возможна трансформация доброкачественной IgM-гаммапатии в макроглобулинемию Вальденстрема, вероятность которой возрастает пропорционально длительности болезни, достигая 27 % к 20-му году болезни [4]. Сообщалось также о возможности развития на поздних этапах болезни лимфоплазмоцитарной лимфомы [14], миеломной болезни [15] и В-клеточной лимфомы маргинальной зоны [16]. Опубликованы также наблюдения развития АА-амилоидоза [17] как осложнения СШ.
Таким образом, возможность малигнизации заболевания обусловливает необходимость тщательного наблюдения таких пациентов на протяжении многих лет. Наши пациенты наблюдаются уже 9 и 6 лет соответственно без развития каких-либо осложнений болезни.
Заключение
СШ является очень редкой патологией, которая проявляется большим разнообразием внекожных симптомов, сопровождается различными диагностическими находками, что затрудняет ведение таких пациентов и не всегда позволяет сразу поставить верный диагноз. Среднее время от появления симптомов до установления диагноза СШ составляет 5 лет [4].Такого пациента могут наблюдать множество врачей: от аллерголога, дерматолога и терапевта до ревматолога, гематолога и онколога. Пациент с СШ может в любой момент обратиться к одному из них, поэтому каждый специалист должен знать особенности течения болезни и ее диагностические критерии. При этом важность знания ключевых особенностей СШ обусловлена не только основными проявлениями болезни, но и угрозой трансформации доброкачественной IgM-гаммапатии в макроглобулинемию Вальденстрема, лимфому или другие гематологические заболе-вания.
В наших наблюдениях мы показали, что в одном случае применение пефлоксацина и ритуксимаба позволяет контролировать воспалительную активность заболевания и, возможно, снижает риск отдаленных осложнений, в другом – эффективность элиминационной диеты в устранении кожных симптомов и высокий уровень общего IgE могут указывать на новый механизм в развитии болезни, что в будущем позволит оптимизировать лечение таких пациентов.
